Theory-guided alloy design for ductile magnesium: from atomic descriptors to multicomponent concepts
Magnesium alloys are among the most attractive lightweight structural materials, combining very low density with high specific strength. Their broader technological deployment, however, remains fundamentally constrained by poor room-temperature formability. This limitation originates in the hexagonal close-packed (hcp) crystal structure of Mg, which provides too few easily activated deformation modes to satisfy the von Mises criterion under ambient conditions. While rare-earth (RE) alloying additions—most notably yttrium—have long been known to dramatically enhance ductility, their high cost, supply risk, and environmental footprint motivate the search for alternative, non-RE design strategies.
Pei_2015_New_J._Phys._17_093009.pdf
PDF-Dokument [2.3 MB]
A recent central contribution is the demonstration that ductility-relevant atomic-scale descriptors can be distilled into a compact, physically interpretable screening parameter, enabling rapid, theory-guided alloy prototyping without the need for exhaustive ab initio calculations. The work establishes a rigorous bridge between stacking-fault energetics, fundamental solute properties, and macroscopic mechanical behavior, and then exploits this bridge to identify promising binary and ternary Mg alloy concepts.
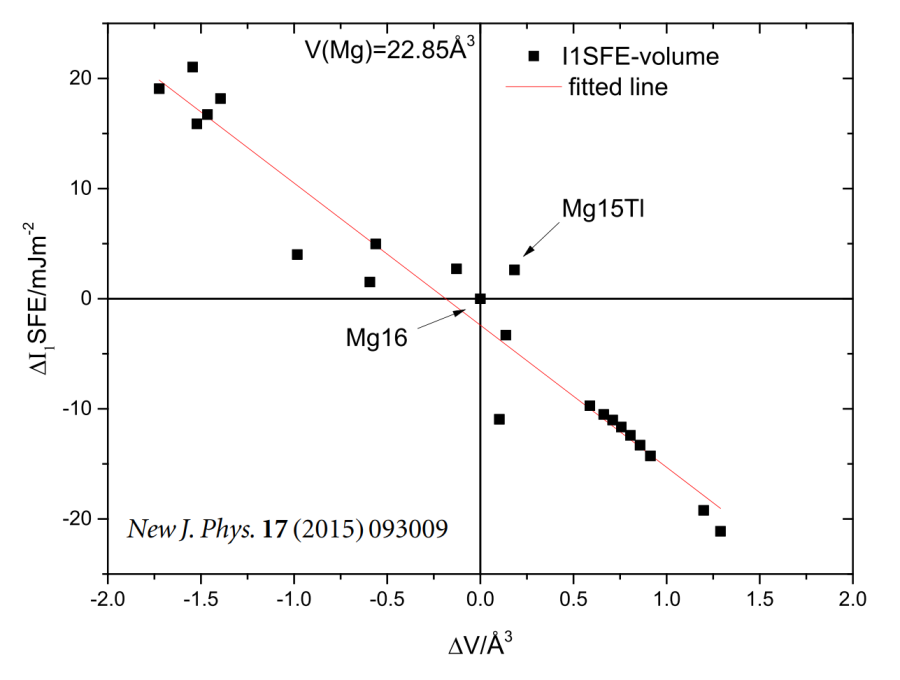 Relative change of the stacking fault energy D I1 SFE of Mg15X as a function of the change of volume per atom in a Mg-alloy D V (Mg15X). The data-points can be divided into two groups by the atomic volume of pure Mg (22.85 Å3). All RE-elements increase
Relative change of the stacking fault energy D I1 SFE of Mg15X as a function of the change of volume per atom in a Mg-alloy D V (Mg15X). The data-points can be divided into two groups by the atomic volume of pure Mg (22.85 Å3). All RE-elements increase
I₁ stacking faults as a scale-bridging descriptor for ductility of Magnesium Alloys
The starting point of the study is the now well-established insight that the I₁ intrinsic stacking fault energy (SFE) in Mg plays a decisive role in enabling non-basal plasticity. Previous combined ab initio and experimental work by the same research group demonstrated that RE additions reduce the I₁ SFE, thereby facilitating the nucleation of non-basal dislocations from I₁ faults and promoting more isotropic plastic flow. In this sense, the I₁ SFE acts as a quantitative atomic-scale proxy for macroscopic ductility.
However, direct quantum-mechanical computation of stacking-fault energies in multicomponent alloys is computationally expensive, particularly for hcp metals where large supercells are required. The present work therefore asks a fundamental question: Can the effect of solutes on I₁ SFE be predicted from simple, tabulated elemental properties?
Using density-functional theory (DFT) within the PAW–GGA framework and the axial next-nearest-neighbor Ising (ANNNI) model, the authors compute I₁ SFEs for 18 binary Mg₁₅X alloys (6.25 at.% solute), including eight RE and ten non-RE elements. These data are then correlated with five candidate descriptors of the solute elements.
Three of these emerge as dominant and physically meaningful predictors:
-
Atomic volume
The I₁ SFE exhibits a strong anti-correlation with the atomic volume of the alloy (Pearson r = −0.97). Larger solutes expand the local lattice, reduce the energetic penalty for faulted stacking, and thereby stabilize I₁ faults. All RE elements fall into this category, whereas most non-RE elements contract the lattice and increase the SFE. -
Electronegativity
A remarkably strong positive correlation (r = 0.95) is observed between solute electronegativity and I₁ SFE. Elements with electronegativity close to Mg (≈ 1.1–1.3 on the Pauling scale) tend to preserve metallic bonding character and reduce fault energies, whereas more electronegative solutes stiffen bonding and raise the SFE. -
Bulk modulus
The I₁ SFE increases with increasing solute bulk modulus, following an approximately logarithmic relationship (r ≈ 0.90). Soft solutes with bulk moduli comparable to Mg (≈ 32–56 GPa) are therefore favorable for ductilization.
Importantly, these three parameters span structural (volume), electronic (electronegativity), and elastic (bulk modulus) aspects of bonding. Their simultaneous relevance highlights the intrinsically multi-physics nature of stacking-fault energetics and mirrors similar correlations previously identified in Fe-, Ni-, and nitride-based alloy systems.
The Yttrium Similarity Index (YSI) for Ductile Magnesium Alloys: a compact descriptor for ductilizing solutes
To operationalize these insights for rapid alloy screening, Pei et al. introduce the Yttrium Similarity Index (YSI). This scalar metric quantifies the “distance” of any solute element from yttrium in a three-dimensional property space spanned by atomic volume, electronegativity, and bulk modulus. Each component is weighted according to its correlation strength with I₁ SFE, and the index is normalized such that YSI = 1 corresponds to yttrium.
When evaluated for 76 elements across the periodic table, the YSI reproduces known trends with striking fidelity:
-
All lanthanides cluster near YSI ≈ 0.95–1.0 and are correctly identified as strong I₁-SFE reducers.
-
Only one non-RE element, lithium, exceeds the critical threshold (YSI ≈ 0.84) required to reduce the I₁ SFE below that of pure Mg.
-
Most technologically common alloying elements (Al, Zn, transition metals) fall well below this threshold, explaining their limited ductilization capability in binary Mg alloys.
The relationship between I₁ SFE and YSI follows an exponential trend, allowing the identification of a quantitative threshold: only solutes with YSI ≳ 0.84 are predicted to reduce the I₁ SFE relative to Mg. This result reveals a fundamental limitation of binary alloying strategies and explains why RE-free ductile Mg alloys are so difficult to realize via single-solute additions.
From binaries to ternaries: overcoming intrinsic limitations
Recognizing this constraint, the authors extend the YSI concept to ternary Mg alloys. The key assumption—well justified at dilute concentrations—is that the effective solute properties can be approximated by arithmetic averages of the individual solute descriptors. This enables the definition of a ternary YSI for any solute pair without performing new DFT calculations.
Out of 2850 non-RE solute pairs, 133 combinations exceed YSI = 0.85, and 11 exceed 0.95. These high-YSI pairs are dominated by combinations involving Ca, Na, Al, Zr, Sr, and Tl, elements that individually are insufficient but collectively can emulate the effect of yttrium in property space.
To validate this approach, the authors perform explicit DFT-ANNNI calculations for 11 selected ternary Mg alloys. In all cases, the predicted solute pairs reduce the I₁ SFE relative to pure Mg, with several combinations—such as (Zr,Ca), (Ti,Ca), (Sr,Al), and (Hf,Ca)—showing very strong reductions, in some cases even yielding negative I₁ SFEs. The 100 % success rate of these predictions represents a remarkable validation of the YSI concept.
With this approach this work establishes a new paradigm for alloy development in hcp metals: rather than searching empirically or relying on brute-force quantum-mechanical screening, one can exploit physically grounded, low-dimensional descriptors to guide materials design. The YSI provides a rare example of a descriptor that is:
-
quantitatively predictive,
-
physically interpretable,
-
computationally inexpensive, and
-
invertible, enabling targeted alloy design.
For magnesium metallurgy, the implications are profound. The results explain why RE elements are uniquely effective, why most conventional alloying strategies fail, and how carefully chosen multicomponent systems can overcome these limitations. Beyond Mg, the methodology exemplifies how electronic structure, elasticity, and crystallography can be fused into actionable design rules, aligning closely with modern concepts of data-driven and sustainability-oriented materials science.
In the broader context of lightweight and sustainable structural materials, Pei et al. provide a blueprint for theory-guided prototyping, bridging atomic-scale physics and engineering-scale performance with exceptional clarity and rigor.
Reference
Pei, Z., Friák, M., Sandlöbes, S., Nazarov, R., Svendsen, B., Raabe, D., Neugebauer, J., Rapid theory-guided prototyping of ductile Mg alloys: from binary to multi-component
materials, New J. Phys. 17 (2015) 093009.