Solid State Green Steelmaking is Governed by a Displaced Reaction-Transformation
We reveal the in-operando reduction dynamics of iron oxide from 200 °C–1000 °C using time-resolved hydrogen-environmental scanning transmission electron microscopy
(3 Pa), bulk analysis, and atomistic modeling. We propose a general cation-mediated redox mechanism in $\mathrm{Fe_3O_4}$: cation diffusion triggers a spatially displaced reaction–transformation,
driving phase changes well beyond the reaction front. At 727 °C, this displacement exceeds previously assumed reaction-front thicknesses by 2–4 orders of magnitude. Our results highlight the
universal, pivotal role of cation diffusion in rapid hydrogen-based metal oxide reduction, providing fundamental atomic-scale insights for sustainable steelmaking, batteries, and
corrosion.
 Solid State Green Steelmaking is Governed by a Displaced Reaction-Transformation : https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.76583
Solid State Green Steelmaking is Governed by a Displaced Reaction-Transformation : https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.76583
Cation Diffusion-Mediated Displaced Reaction-Transformation in Green Steelmaking
Advanced Science , 2026; 0:e76583
Advanced Science - 2026 - Cation Diffusi[...]
PDF-Dokument [4.1 MB]
Advanced Science , 2026; 0:e76583
Advanced Science - 2026 - Cation Diffusi[...]
PDF-Dokument [4.1 MB]
Hydrogen-based direct reduction of iron oxides shows great potential for decarbonizing the iron and steelmaking industry. However, deciphering such complex,
multistage reactions remains challenging due to the concurrent occurrence of multiple nonlinearly coupled processes. Here, the in-operando reduction dynamics of iron oxide ranging from 200 °C–1000 °C
is revealed using time-resolved hydrogen-environmental scanning transmission electron microscopy (with a hydrogen gas pressure of 3 Pa) combined with bulk analysis and atomistic modeling. Proposing a
general cation-mediated redox mechanism in Fe3O4, cation diffusion triggers a spatially displaced reaction–transformation and drives the ensuing dynamic phase transformations and reduction steps that
extend significantly beyond the reaction front. At 727 °C the spatial displacement is 2–4 orders of magnitude above the previously assumed reaction-front thickness. The results underscore the
cross-scale universality and pivotal role of cation diffusion in enabling rapid hydrogen-based reduction of metal oxides, offering not only fundamental atomic-scale insights to help render the
steelmaking industry more sustainable, but also a better understanding of related processes in batteries and corrosion.
 Dynamic surface topological reconstruction via a ledge growth mechanism. (a) A general concept of the displaced reaction-transformation that occurs when the diffusion rate outperforms the reaction rate in an individual reaction. Here, blue and green spher
Dynamic surface topological reconstruction via a ledge growth mechanism. (a) A general concept of the displaced reaction-transformation that occurs when the diffusion rate outperforms the reaction rate in an individual reaction. Here, blue and green spher
Details about the Displaced Reaction-Transformation in Iron Oxide Reduction
Decarbonizing the steelmaking industry requires a fundamental understanding of hydrogen-based direct reduction (HyDR) kinetics in iron oxides. Deciphering these
complex reactions demands moving beyond macroscopic observations to track the local solid-state transport that connects gas-solid interactions with the actual migration of phase transformation
fronts.
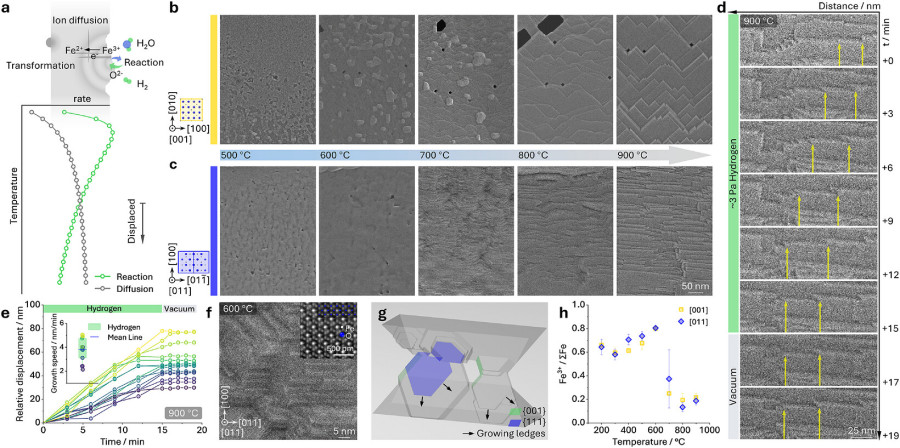
Through time-resolved hydrogen-environmental scanning transmission electron microscopy operated at 3 Pa H2 across 200°C to 1000°C, combined with atomistic modeling,
a decoupled redox mechanism emerges. When the migration of Fe cations outpaces the local surface reduction reaction, phase transformation sites physically decouple from the initial reduction front.
This process, termed a displaced reaction-transformation, operates at a spatial displacement two to four orders of magnitude larger than previously assumed reaction-front thicknesses.
This mechanism is strictly governed by rapid interstitial Fe cation diffusion. As H2 strips oxygen from the non-polar {100} surfaces of the Fe3O4 (magnetite)
lattice, excess interstitial Fe cations accumulate. Driven by a steep chemical potential gradient, these highly mobile cations undergo long-range diffusion toward O-rich, polarized {111} planes. This
directed mass transport triggers a dynamic surface topological reconstruction, manifesting as a ledge-controlled growth mechanism on the {111} planes, tightly coupled with the phase transformation to
Fe(1-x)O. At 900°C, this directional ledge growth proceeds at approximately 3.8 nm per minute and halts immediately when the H2 supply is removed, proving it is a reaction-driven, rather than purely
thermal, process.
These experimental observations are rigorously corroborated by atomic cluster expansion (ACE) interatomic potential simulations. The models confirm that
interstitial Fe cations preferentially diffuse to O-rich Fe3O4 (magnetite) {111} surfaces, facilitating the thermodynamically favorable nucleation and growth
of FeO {111} facets over additional Fe3O4 (magnetite) layers.
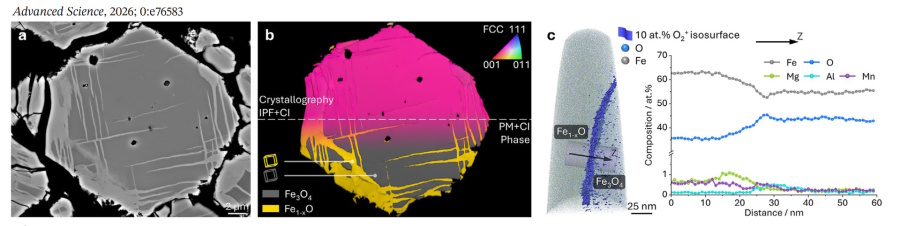
Crucially, this nanoscale mechanism scales directly to bulk thermodynamic processes. In microscale $Fe_{3}O_{4}$ ore reduced at 900°C under $10^{5}$ Pa $H_{2}$,
accelerated cation diffusion drives the phase transformation tens of micrometers deep into the bulk. This yields a three-dimensional net-plate-like distribution of $Fe_{1-x}O$ inclusions penetrating
the $Fe_{3}O_{4}$ matrix, rather than a uniform surface shell.
Cation-mediated displaced reaction-transformation constitutes a cross-scale, universal mechanism governing the direct reduction of Fe3O4
(magnetite), and it is likely applicable to other reducible spinel-structured transition metal oxides. Modulating these cation transport networks—via grain boundary
engineering, dislocation density control, or targeted surface faceting—provides a deterministic kinetic lever to optimize structural evolution in green steelmaking, with analogous implications for
degradation in corrosion and solid-state transport in battery systems.
![Ledge growth mechanism and interstitial Fe cation diffusion. (a,b) Sketch of the ledge growth mechanism of surface terraces observed during in situ HyDR experiments on (a) the [001]-oriented and (b) the [011]-oriented Fe3O4 samples (see Figure 2b). Surfac](https://www.dierk-raabe.com/s/cc_images/teaserbox_2498772773.jpg?t=1784214107) Ledge growth mechanism and interstitial Fe cation diffusion. (a,b) Sketch of the ledge growth mechanism of surface terraces observed during in situ HyDR experiments on (a) the [001]-oriented and (b) the [011]-oriented Fe3O4 samples (see Figure 2b). Surfac
Ledge growth mechanism and interstitial Fe cation diffusion. (a,b) Sketch of the ledge growth mechanism of surface terraces observed during in situ HyDR experiments on (a) the [001]-oriented and (b) the [011]-oriented Fe3O4 samples (see Figure 2b). Surfac