Development of an atomic cluster expansion potential for iron and its oxides
The combined structural and electronic complexity of iron oxides poses many challenges to atomistic
modeling. To leverage limitations in terms of the accessible length and time scales, one requires a
physically justifi ed interatomic potential which is accurate to correctly account for the complexity of
iron-oxygen systems. Such a potential is not yet available in the literature. In this work, we propose a
machine-learning potential based on the Atomic Cluster Expansion for modeling the iron-oxygen
system, which explicitly accounts for magnetism. We test the potential on a wide range of properties of
iron and its oxides, and demonstrate its ability to describe the thermodynamics of systems spanning
the whole range of oxygen content and including magnetic degrees of freedom.
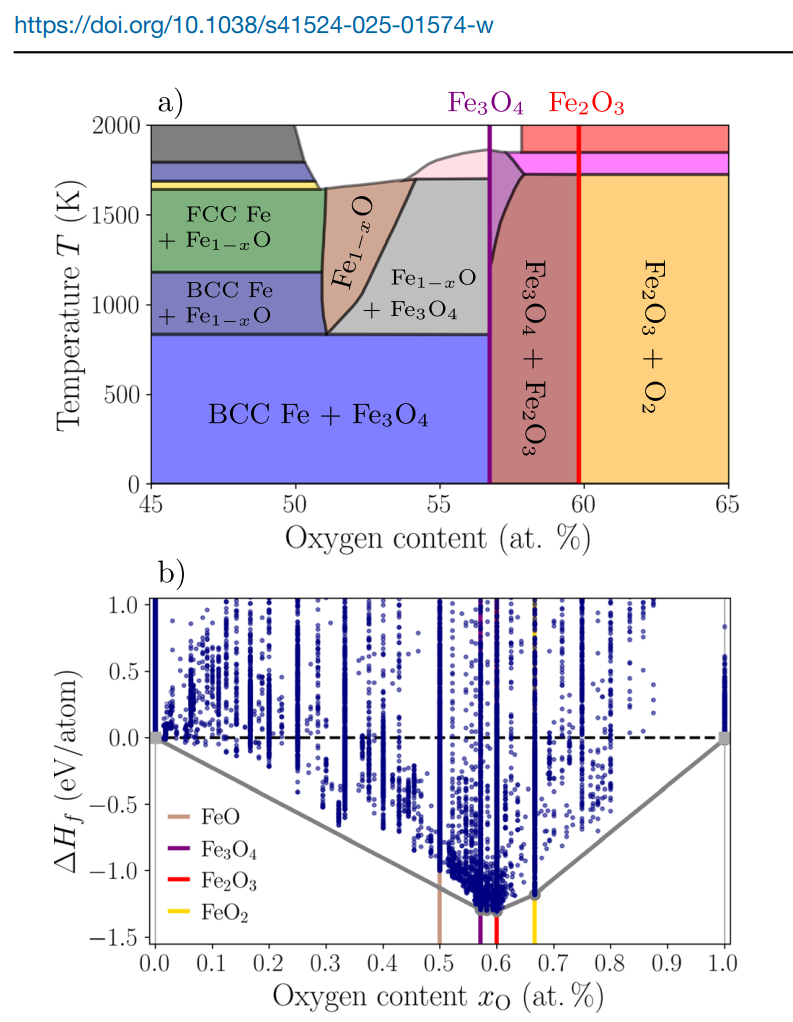 Development of an atomic cluster expansion potential for iron and its oxides: Experimental phase diagram and DFT reference dataset. a Experimental Fe-O phase diagram adapted from ref. 8 showing the stable compounds. b DFT convex Hull (i.e., formation enth
Development of an atomic cluster expansion potential for iron and its oxides: Experimental phase diagram and DFT reference dataset. a Experimental Fe-O phase diagram adapted from ref. 8 showing the stable compounds. b DFT convex Hull (i.e., formation enth
Atomic cluster expansion potential for iron and its oxides
The combined structural and electronic complexity of iron oxides poses many challenges to atomistic modeling. To leverage limitations in terms of the accessible length and time scales, one requires a physically justifi ed interatomic potential which is accurate to correctly account for the complexity of iron-oxygen systems. Such a potential is not yet available in the literature. In this work, we propose a machine-learning potential based on the Atomic Cluster Expansion for modeling the iron-oxygen system, which explicitly accounts for magnetism. We test the potential on a wide range of properties of iron and its oxides, and demonstrate its ability to describe the thermodynamics of systems spanning the whole range of oxygen content and including magnetic degrees of freedom.
Development of an atomic cluster expansi[...]
PDF-Dokument [2.2 MB]
The combined structural and electronic complexity of iron oxides poses many challenges to atomistic modeling. To leverage limitations in terms of the accessible length and time scales, one requires a physically justifi ed interatomic potential which is accurate to correctly account for the complexity of iron-oxygen systems. Such a potential is not yet available in the literature. In this work, we propose a machine-learning potential based on the Atomic Cluster Expansion for modeling the iron-oxygen system, which explicitly accounts for magnetism. We test the potential on a wide range of properties of iron and its oxides, and demonstrate its ability to describe the thermodynamics of systems spanning the whole range of oxygen content and including magnetic degrees of freedom.
Development of an atomic cluster expansi[...]
PDF-Dokument [2.2 MB]
Iron is one of Earth's most abundant elements and the foundation of steels and metallic alloys. Naturally occurring in oxide form, it requires a deep understanding
of iron oxide properties at the atomic and electronic levels to optimize iron production and minimize oxidation-related degradation.
The Fe-O binary system, though containing few stable compounds, shows remarkable structural and electronic complexity. Iron oxides appear in nature as three stable
minerals: wüstite (FeO), magnetite (Fe₃O₄), and hematite (Fe₂O₃), ordered by increasing oxygen concentration.
Most atomistic studies of Fe-O rely on Density Functional Theory (DFT). However, accurately describing these compounds requires different exchange-correlation
functionals depending on oxygen content. Standard semi-local DFT functionals incorrectly predict iron oxides to be metallic, prompting the use of more sophisticated approaches like DFT+U or hybrid
functionals.
First-principles calculations are limited in system size and simulation timescales, making them impractical for studying extended defects, diffusion, or phase
transformations. These complex phenomena require interatomic potentials that are both accurate and efficient — a significant challenge for the Fe-O system across varying temperatures,
stoichiometries, and conditions.
Existing Fe-O interatomic potentials, mostly based on ReaxFF, show considerable discrepancies with DFT and experimental data. Notably, several models predict all
three iron oxides to be dynamically unstable, while a recently developed analytical bond order potential struggles with higher oxygen-content phases.
In this work, we develop a machine-learned interatomic potential (MLIP) based on atomic cluster expansion (ACE), explicitly incorporating magnetism for a physically
accurate description of the Fe-O system. Fitted to an extensive DFT database, the potential is validated across pristine and defective phases and demonstrates accurate magnetic and finite-temperature
behavior.