Compositionally complex and High Entropy Alloys via physics-informed artificial intelligence
We are, by any reasonable estimate, still at the very beginning of materials discovery. Of the roughly 10⁸⁰ chemically meaningful multi-element combinations that we could in principle synthesize, we use on the order of 10⁴ materials industrially today. We see this gap as the central motivation for pushing beyond binary and ternary alloy design into compositionally complex systems — high-entropy alloys, multi-principal-element ceramics, chemically graded superalloy microstructures, and near-atomic-scale-blended semiconductor stacks. When we add elements to a solid solution, we do far more than shift a point on a phase diagram: we perturb the electronic density of states, introduce or suppress magnetic ordering, break local crystal symmetry, generate short-range order, and — this is the part we find most consequential for engineering properties — we change the formation energies, migration barriers and binding behavior of point defects, dislocations and interfaces, often exponentially with composition. Because materials are essentially never used in thermodynamic equilibrium but in metastable, defect-populated states, we cannot separate the question of chemical complexity from the question of microstructural complexity: one drives the other.
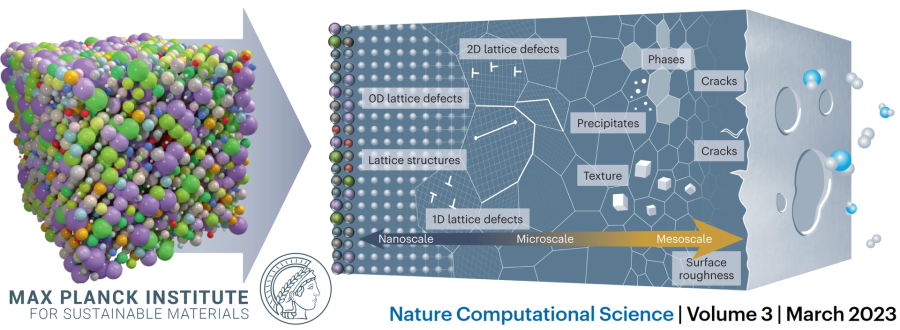
Where our physics-based toolbox reaches its limits
We rely on density functional theory (DFT) as our most rigorous quantum-mechanical tool for extracting energetics, but we are constrained by supercell sizes that are far too small to sample the
configurational space of a five- or six-component random solid solution with proper statistical representation of local chemical environments. We do not expect quantum computing to resolve this
bottleneck for large, chemically complex systems in the near term. When we move to molecular dynamics with machine-learning-trained interatomic potentials, we can reach larger system sizes and
include kinetics, but we still depend on how faithfully the potential reproduces the true multi-body energy landscape. At the mesoscale, we use phase-field and crystal-plasticity frameworks to couple
chemistry, elasticity and diffusion, but these methods rely on coarse-grained Landau-type free-energy approximations whose fidelity is only as good as the underlying thermodynamic description.
We find a particularly instructive failure mode in the Cantor-type FeMnCrNiCo family. This alloy combines some of the strongest ferromagnetic (Fe, Co, Ni) and antiferromagnetic (Mn, Cr) elements
within a single metastable solid solution, so we must simultaneously capture the correct magnetic ground states across all relevant spin configurations and properly account for configurational,
vibrational, magnetic and electronic entropy contributions — not just the configurational term that dominates simple mixing models. Most empirical potentials do not have the functional flexibility to
do this, and as a result even well-established bottom-up atomistic approaches fail to reproduce known experimental phase boundaries in these systems. We see an analogous, and at first
counterintuitive, phenomenon in high-entropy ceramics: despite their strong covalent/ionic bonding and localized electrons — a chemistry we would traditionally expect to be enthalpy-dominated —
disorder-induced charge fluctuations and configurational entropy on the anion sublattice open up mixing behavior and compositional space that we would not predict from classical ionic-bonding
arguments alone.
Given these constraints, we often fall back on physics-based descriptors: computationally tractable quantities with high leverage on the properties we care about. A recurring example in our own and
related work is the use of DFT-computed stacking-fault energy as a proxy for phase stability, deformation-twinning propensity and martensitic transformability in high-entropy alloys. We couple such
descriptors to crystal-plasticity or phase-field solvers to predict mechanical response, and this strategy has enabled us to design alloys such as metastable dual-phase high-entropy systems in which
two coexisting phases undergo complementary, load-triggered transformation-induced plasticity and twinning-induced plasticity mechanisms. What we cannot yet do with descriptor-based approaches is
resolve the full energetic-kinetic-microstructure-property chain in a single, generalizable framework — each descriptor is tied to the specific mechanism we identified as rate-limiting for that
particular problem.
Table: Complexity of materials with multiple principal elements (for example, high-entropy alloys) or impurity elements (for example, recycled alloys) and the role of computational materials science
|
Challenges in materials design |
Computational challenges and opportunities specific to multi-component and chemically complex materials |
|---|---|
|
Characteristic bulk features • Up to ten alloying and/or impurity elements • Multiple (meta-)stable phases and phase transformations • Crystal and amorphous structures • Strong distortions and local crystalline symmetry-breaking • Chemical ordering • Segregation and heterogeneity across all scales • Challenging synthesis, fabrication and processing • Near-equilibrium or highly non-equilibrium synthesis pathways |
Challenges • Prediction of bulk phase energies and kinetics • Prediction of short-range ordering • Insufficient databases and training data, that is, how to do AI with sparse data • Electronic and magnetic effects • Configurational, vibrational, magnetic and electronic entropy • Bridging discrete and continuum models • Macroscopic response from electronic and atomistic principles Opportunities • Extraction of thermodynamic data from literature by AI • Hybrid machine learning and active-learning methods for sparse data problems • AI for bridging scales and accelerated solvers • AI for bridging atomistic and continuum simulations • AI-guided automatic and even autonomous high-throughput simulations |
|
Lattice defects • Solute-decorated point defects, line defects, interfaces and so on • Microstructure patterning and gradients |
Challenges • Energies and kinetics of defects in chemical multi-component decorated state (due to the Gibbs adsorption isotherm) • From individual solute-decorated defects to patterning and gradients • Multi-physics interactions: chemistry, magnetism, mechanics • Process and property simulation Opportunities • Surrogating defect features and patterning with AI • Structure–chemistry–process–property linkage through AI • New routes for stabilizing nanocrystalline structures and chemically decorated defects |
|
Life cycle, longevity, sustainability • Considering element scarcity, mining, refining, synthesis, processing and recycling • Corrosion and longevity in harsh environments |
Challenges • Considering sustainability, material decay, harsh environments, element scarcity and life-cycle aspects in the design of chemically complex materials for a circular economy • Reconciliation of material design for advanced properties with the goal of improving material sustainability Opportunities • Holistic cradle-to-grave simulations and AI methods for materials, processes and recycling • High-component surface oxides and nitrides with better corrosion resistance • New materials with enhanced abrasion, fatigue and stress-corrosion resistance |
|
Increased amount of data • Materials evolved in part over millennia • Materials design often done via trial and error • Dispersed knowledge (literature, patents, experience, communities) |
Challenges • Systematic and quality-controlled collection of knowledge • No homogeneous or unified nomenclature, materials, processes and compositions Opportunities • Autonomous AI-based knowledge extraction from literature • Autonomous AI-based material and process design • AI guidance for material development |
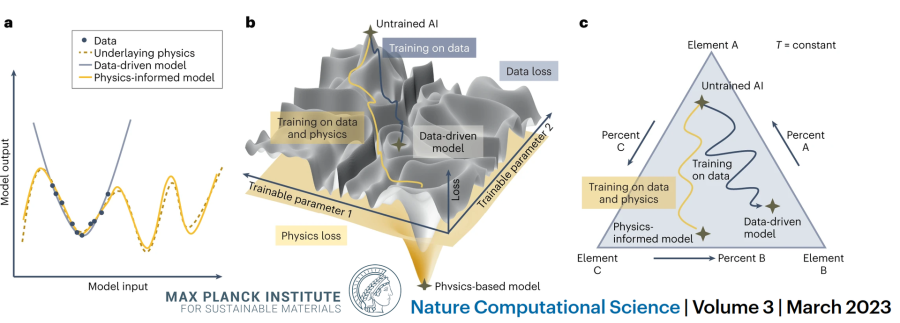 a, Example case where a limited number of sample data (black dots) from an underlying physical phenomenon (dashed line) are used to train an artificial neural network model. The model can be trained only on the data (gray solid line) or in a hybrid method
a, Example case where a limited number of sample data (black dots) from an underlying physical phenomenon (dashed line) are used to train an artificial neural network model. The model can be trained only on the data (gray solid line) or in a hybrid method
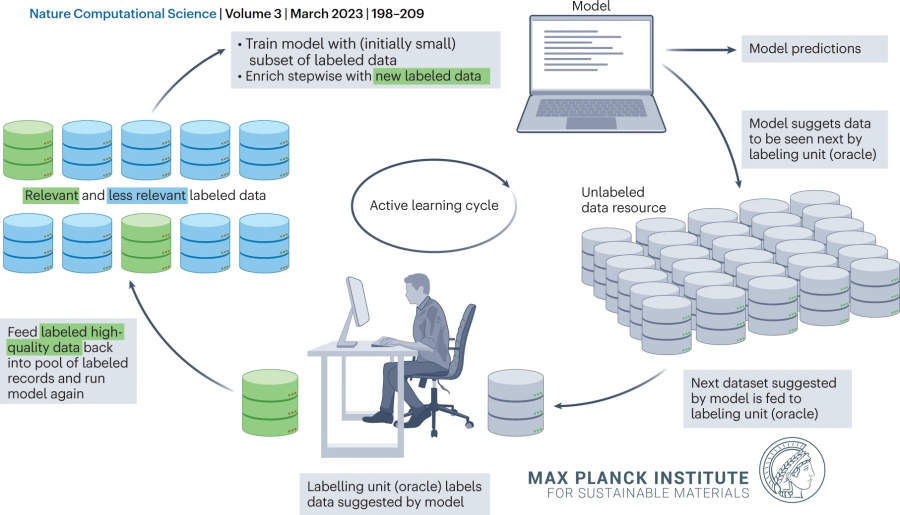 Active learning is a powerful approach in which a key problem of AI, namely, the challenge of a limited availability of well-labeled data (one of the most expensive steps in AI) for training is tackled. This is done through iterative machine-assisted deci
Active learning is a powerful approach in which a key problem of AI, namely, the challenge of a limited availability of well-labeled data (one of the most expensive steps in AI) for training is tackled. This is done through iterative machine-assisted deci
AI as a dimensionality-reduction strategy in alloy design, not a shortcut
- We use an iterative active-learning loop — coupling machine learning, AI-guided experiments and DFT-based phase-equilibrium calculations — to identify new FeNiCo-based Invar compositions (alloys with near-zero thermal expansion), closing the loop between thermodynamic simulation and targeted experimental validation rather than screening blindly.
- We apply symbolic regression, guided by sure-independence-screening and sparsifying-operator approaches, to derive closed-form analytical expressions linking lattice constant and cohesive energy to the bulk modulus of perovskites — recovering physically interpretable descriptors rather than an opaque regression surface.
- We use random-forest models trained against DFT-computed entropy-forming-ability metrics to screen multi-component disordered metal carbides, from which we flagged around 70 new candidate high-entropy ceramic compositions for further validation.
- We have shown, in the case of nickel-base superalloys, that a neural network trained on experimental and CALPHAD-derived thermodynamic data can optimize compositions across strength, corrosion resistance and phase stability simultaneously — a multi-objective, high-dimensional design problem that we consider essentially intractable for forward physics models alone, given the coupling between processing history, γ/γ′ microstructure and mechanical/oxidation response