Metallurgical Alloy Design
What is alloy design and why do we do it?
With the term alloy design we refer to the knowledge-guided approach for the development and compositionally sensitive design of novel metallurgical materials.
In the field of engineering we are typically using only up to 1000 different types of metallic alloys on a regular basis.
This is a very tiny fraction of potentially interesting alloys. This also means that the identification of new compositions with correspondingly suited properties and microstructures can not be
readily pursued by using conventional empirical and stepwise approaches. Of course, in case of having identified beneficial alloys we still have to conduct careful iteration to optimize
properties, however, this is not a suited approach when trying to screen and identify entirely new alloy compositions with so far unprecedented property combinations.
For this reason we must develop and utilize more systematic approaches for identifying metallurgical treasure maps that help us to identify suited compositions and microstructures for new alloy
regimes.
The systematics can come from computational materials science, the use of semi-empirical chemical rules pertaining to the underlying electronic bonds and structure motifs or from combinatorial
metallurgical synthesis. Other approaches are to reflect which type of property we actually aim to design and from that starting point on identify the corresponding key parameters and optimize
their associated underlying thermodynamic or kinetic quantities such as certain dislocation properties, certain interface properties or certain phase transformation properties.
Alloy design for density reduced TWIP steels
We introduce a new experimental approach to the compositional and thermo-mechanical design and rapid maturation of bulk structural materials. This method, termed rapid alloy prototyping
(RAP), is based on semi-continuous high throughput bulk casting, rolling, heat treatment and sample preparation techniques. 45 Material conditions, i.e. 5 alloys with systematically varied
compositions, each modified by 9 different ageing treatments, were produced and investigated within 35 h. This accelerated screening of the tensile, hardness and microstructural properties
as a function of chemical and thermo-mechanical parameters allows the highly efficient and knowledgebased
design of bulk structural alloys. The efficiency of the approach was demonstrated on a group of Fe–30Mn–1.2C–xAl steels which exhibit a wide spectrum of structural and mechanical
characteristics, depending on the respective Al concentration. High amounts of Al addition (>8 wt.%) resulted in pronounced strengthening, while low concentrations (<2 wt.%) led to
embrittlement of the material during ageing.
 TWIP steel with optimized stacking fault energy.
TWIP steel with optimized stacking fault energy.
Acta Materialia 60 (2012) 4950–4959
Acta Materialia 60 (2012) 4950 combinato[...]
PDF-Dokument [1.3 MB]
Alloy design and combinatorial synthesis for weight-reduced Fe-Mn-Al-C austenitic TWIP steels
We present recent developments in the field of austenitic steels with up to 18%
reduced mass density. The alloys are based on the Fe-Mn-Al-C system. Here,
two steel types are addressed. The first one is a class of low-density twinning-induced plasticity or single phase austenitic TWIP (SIMPLEX) steels with
25–30 wt.% Mn and<4–5 wt.% Al or even<8 wt.% Al when naturally aged.
The second one is a class of j-carbide strengthened austenitic steels with even
higher Al content. Here, j-carbides form either at 500–600C or even during
quenching for >10 wt.% Al. Three topics are addressed in more detail,
namely, the combinatorial bulk high-throughput design of a wide range of
corresponding alloy variants, the development of microstructure–property
relations for such steels, and their susceptibility to hydrogen embrittlement.
 Combinatorial metallurgical synthesis applied to TWIP steels and weight reduced steels.
Combinatorial metallurgical synthesis applied to TWIP steels and weight reduced steels.
JOM, Vol. 66, No. 9, 2014
JOM, Vol. 66, No. 9, 2014 page 1845 Low-[...]
PDF-Dokument [3.2 MB]
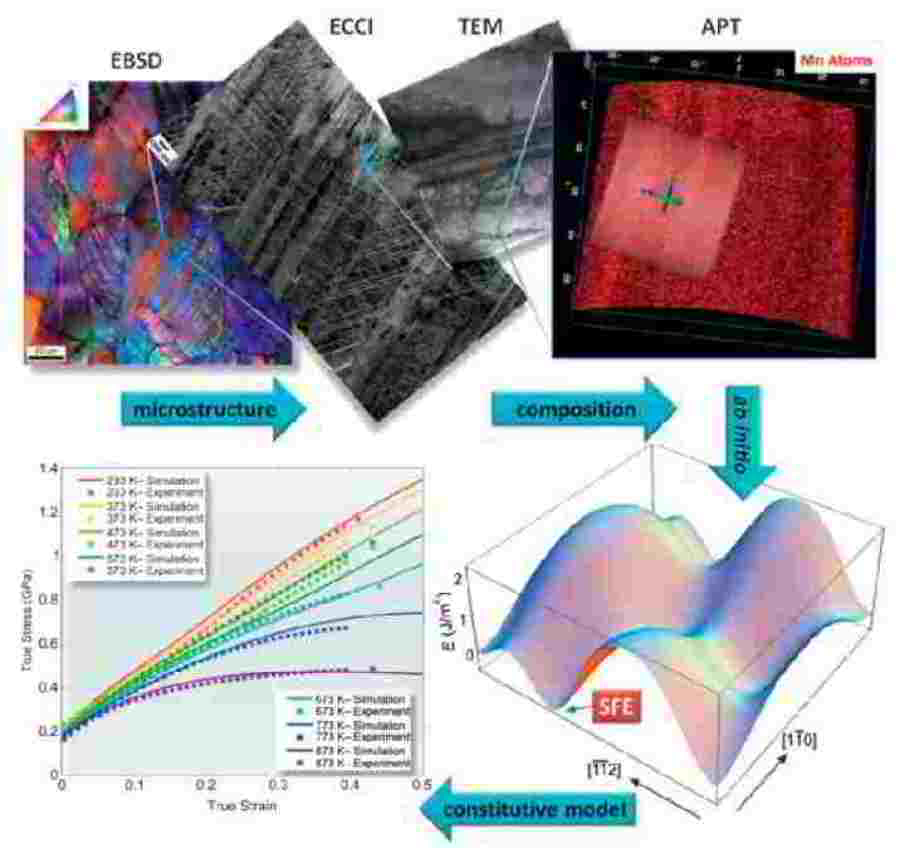
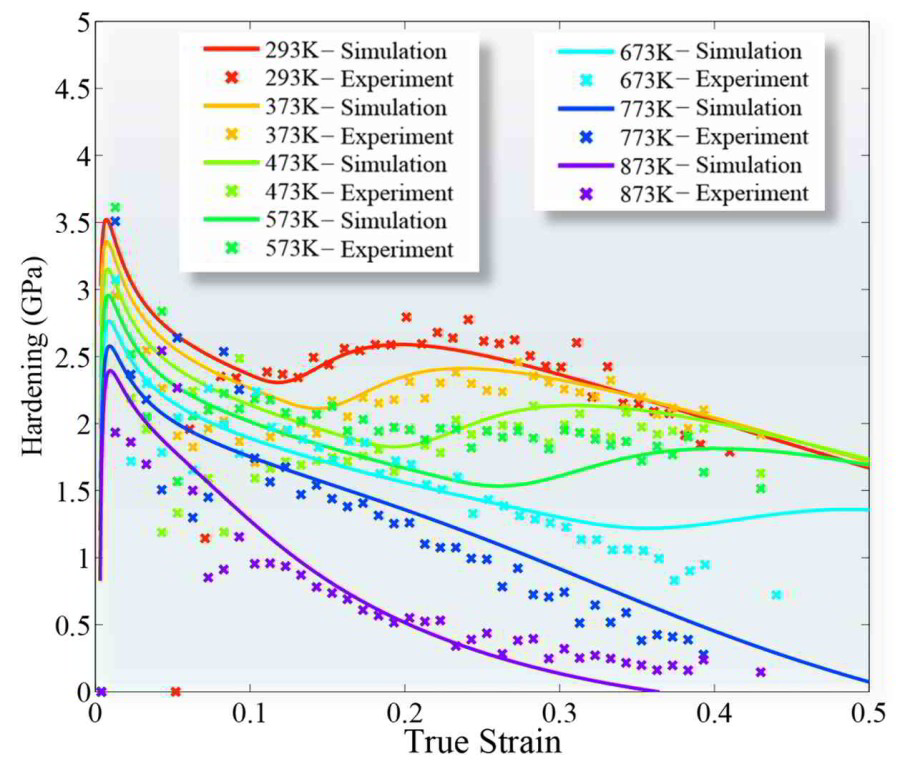
Theory-guided alloy design of twinning induced plasticity steels
We present a multiscale dislocation density-based constitutive model for the strain-hardening behavior in twinning-induced plasticity (TWIP) steels. The approach is a physics-based strain
rate- and temperature-sensitive model which reflects microstructural investigations of twins and dislocation structures in TWIP steels. One distinct advantage of the approach is that the model
parameters, some of which are derived by ab initio predictions, are physics-based and known within an order of magnitude. This allows more complex microstructural information to be included
in the model without losing the ability to identify reasonable initial values and bounds for all parameters.
Dislocation cells, grain size and twin volume fraction evolution are included. Particular attention is placed on the mechanism by which new deformation twins are nucleated, and a new formulation
for the critical twinning stress is presented. Various temperatures were included in the parameter optimization process. Dissipative heating is also considered. The use of physically justified
parameters enables the identification of a universal parameter set for the example of an Fe–22Mn–0.6C TWIP steel.
 Theory-guided alloy design of twinning induced plasticity steels.
Theory-guided alloy design of twinning induced plasticity steels.
 Theory-guided alloy design of twinning induced plasticity steels
Theory-guided alloy design of twinning induced plasticity steels
Acta Materialia 61 (2013) 494-510
Acta Materialia 61 (2013) 494 ab initio [...]
PDF-Dokument [1.8 MB]
Theory-guided design of elastic properties of twinning induced plasticity steels
We have studied experimentally and theoretically the influence of C and Mn content on the Young’s modulus of Fe–Mn–C alloys. Combinatorial thin film and bulk samples were characterized
regarding their structure, texture and Young’s modulus. The following chemical composition range was investigated: 1.5–3.0 at.% C, 28.0–37.5 at.% Mn and 60.6–69.8 at.% Fe. The experimental
lattice parameters change marginally within 3.597–3.614A ˚ with the addition of C and are consistent with ab initio calculations. The Young’s modulus
data are in the range of 185 ± 12–251 ± 59 GPa for the bulk samples and the thin film, respectively. C has no significant effect on the Young’s modulus of these alloys within the composition
range studied here. The ab initio calculations are 15–22% larger than the average Young’s modulus values of the as-deposited and polished thin film at 3 at.% C. The comparison of thin film and
bulk samples results reveals similar elastic properties for equivalent compositions, indicating that the applied research strategy consisting of the combinatorial thin film approach in
conjunction with ab initio calculations is useful to study the composition dependence of the structure and elastic
properties of Fe–Mn–C alloys. The very good agreement between the presented calculations and the experimentally determined lattice parameters and Young’s modulus values implies that the
here-adopted simulation strategy yields a reliable description of carbon in Fe–Mn alloys, important for future alloy design.
Acta Materialia 60 (2012) 6025-6032
Acta Materialia 60 (2012) 6025–6032 Comb[...]
PDF-Dokument [887.1 KB]
Metallic Glasses
Revealing the relationships between chemistry, topology and stiffness of ultrastrong Co-based metallic glass thin films: A combinatorial approach
Acta Materialia 107 (2016) 213-219
Acta Materialia 107 (2016) 213 combinato[...]
PDF-Dokument [2.3 MB]
An efficient way to study the relationship between chemical composition and mechanical properties of thin films is to utilize the combinatorial approach, where spatially resolved mechanical
property measurements are conducted along a concentration gradient. However, for thin film glasses many properties
including the mechanical response are affected by chemical topology. Here a novel method is introduced which enables spatially resolved short range order analysis along concentration gradients
of combinatorially synthesized metallic glass thin films. For this purpose a CoZrTaB metallic glass film of 3 mm
thickness is deposited on a polyimide foil, which is investigated by high energy X-ray diffraction in transmission mode. Through the correlative chemistry-topology-stiffness investigation, we
observe that an increase in metalloid concentration from 26.4 to 32.7 at% and the associated formation of localized
(hybridized) metal e metalloid bonds induce a 10% increase in stiffness. Concomitantly, along the same composition gradient, a metalloid-concentration-induced increase in first order metal -
metal bond distances of 1% is observed, which infers itinerant (metallic) bond weakening. Hence, the metalloid
concentration induced increase in hybridized bonding dominates the corresponding weakening of metallic bonds.
High Stiffness Steels - MMC Steels
In-situ metal matrix composite steels: Effect of alloying and annealing on morphology, structure and mechanical properties of TiB2 particle containing high modulus steels
Acta Materialia 107 (2016) 38-48
Acta Materialia 107 (2016) 38 In-situ MM[...]
PDF-Dokument [1.9 MB]
We systematically study the morphology, size and dispersion of TiB2 particles formed in-situ from Fe-TiB2 based melts, as well as their chemical composition, crystal structure and mechanical
properties. The effects of 5 wt.% additions of Cr, Ni, Co, Mo, W, Mn, Al, Si, V, Ta, Nb and Zr, respectively, as well as
additional annealing treatments, were investigated in order to derive guidelines for the knowledge based alloy design of steels with an increased stiffness / density ratio and sufficiently high
ductility. All alloying elements were found to increase the size of the coarse primary TiB2 particles, while Co led to the most
homogeneous size distribution. The size of the eutectic TiB2 constituents was decreased by all alloying additions except Ni, while their aspect ratio was little affected. No clear relation
between chemical composition, crystal structure and mechanical properties of the particles could be observed. Annealing of
the as-cast alloys slightly increased the size of the primary particles, but at the same time strongly spheroidised the eutectics. Additions of Co and Cr appear thus as the best starting point
for designing novel in-situ high modulus metal matrix composite steels, while using Mn in concert with thermomechanical
processing is most suited to adapt the matrix' microstructure and optimise the particle / matrix co-deformation processes.
Acta Materialia 96 (2015) 47-56
Acta Materialia 96 (2015) 47 high modulu[...]
PDF-Dokument [3.0 MB]
Microstructures of Fe–TiB2 metal-matrix-composites formed in-situ from Fe–Ti–B melts were investigated for hypo- and hyper-eutectic concentrations down to atomic-scale resolution. Special
emphasis is laid on the influence of the solidification rate on particle size, morphology and distribution as well
as their relation to mechanical properties. Innovative routes for the cost-effective production of stiff and ductile high modulus steels for lightweight structural applications are discussed,
focusing on hyper-eutectic compositions due to their high stiffness/density ratio: firstly, very slow cooling allows
the primary particles floating to the top of the cast, from which they can either be easily removed for retaining bulk material containing only fine-dispersed eutectic particles, or be kept and
utilised as a wear resistant surface. Secondly, annealing of amorphous matrix material obtained from very fast solidification
leads to fine dispersed nano-scaled precipitation of TiB2 particles.
Acta Materialia 59 (2011) 4653-4664
C. Herrera, D. Ponge, D. Raabe
Design of a novel Mn-based 1 GPa duplex stainless TRIP steel with 60% ductility by a reduction of austenite stability
Acta Materialia 59 (2011) 4653 High Mn h[...]
PDF-Dokument [1.0 MB]
We report on the microstructure, texture and deformation mechanisms of a novel ductile lean duplex stainless steel (Fe–19.9Cr–0.42Ni–0.16N–4.79Mn–0.11C–0.46Cu–0.35Si, wt.%). The austenite is
stabilized by Mn, C, and N (instead of Ni). The microstructure is characterized by electron channeling contrast imaging (ECCI) for dislocation mapping and electron backscattering diffraction
(EBSD)
for texture and phase mapping. The material has 1 GPa ultimate tensile strength and an elongation to fracture of above 60%. The mechanical behavior is interpreted in terms of the strength of
both the starting phases, austenite and ferrite, and the amount, dispersion, and transformation kinetics of the mechanically induced martensite (TRIP effect). Transformation proceeds from
austenite to hexagonal martensite to near cubic martensite (gamma to epsilon to alpha'). The epsilon-martensite forms in the austenite with an orientation relationship close to Shoji–Nishiyama.
The alpha'-martensite nucleates at the intersections of deformation bands, especially e-bands, with Kurdjumov–Sachs and Nishiyama–Wassermann relationships. The ferrite deforms by dislocation
slip and contains cell substructures.
Titanium Alloys
Elastically Soft Beta Titanium Alloys
Acta Materialia 55 (2007) 4475-4487
Acta Materialia 55 (2007) 4475–4487 ab i[...]
PDF-Dokument [1.9 MB]
In this study we present a new strategy for the theory-guided bottom up design of b-Ti alloys for biomedical applications using a quantum mechanical approach in conjunction with experiments.
Parameter-free density functional theory calculations are used to provide theoretical guidance in selecting and optimizing Ti-based alloys with respect to three constraints: (i) the use of
non-toxic alloy elements; (ii) the stabilization of the body centered cubic b-phase at room temperature; (iii) the reduction of the elastic stiffness compared to
existing Ti-based alloys. Following the theoretical predictions, the alloys of interest are cast and characterized with respect to their crystallographic
structure, microstructure, texture, and elastic stiffness. Due to the complexity of the ab initio calculations, the simulations have been focused on a set of binary systems of Ti with two
different high melting body-centered cubic metals, namely, Nb and Mo. Various levels of model approximations to describe mechanical and thermodynamic properties are tested and critically
evaluated.
The experiments are conducted both, on some of the binary alloys and on two more complex engineering alloy variants, namely, Ti–35 wt.% Nb–7 wt.% Zr–5 wt.% Ta and Ti–20 wt.% Mo–7 wt.% Zr–5 wt.%
Ta.
Acta Materialia 97 (2015) 291-304
Z. Tarzimoghadam, S. Sandlöbes, K.G. Pradeep, D. Raabe
Microstructure design and mechanical properties in a near-a Ti-4Mo alloy
Acta Materialia 97 (2015) 291 Ti Mo Micr[...]
PDF-Dokument [3.8 MB]
We study the effects of different heat treatment routes on microstructure engineering and the resulting mechanical response in a plain binary Ti–4Mo (wt%) model alloy. We observe a broad
variety of microstructure formation mechanisms including diffusion driven allotropic phase transformations as
well as shear and/or diffusion dominated modes of martensitic transformations, enabling a wealth of effective microstructure design options even in such a simple binary Ti alloy. This wide
variety of microstructures allows tailoring the mechanical properties ranging from low yield strength (350 MPa) and high ductility (30–35% tensile elongation) to very high yield strength
(1100 MPa) and medium ductility (10–15% tensile elongation) as well as a variety of intermediate states. Mechanical testing and microstructure characterization using optical microscopy, scanning
electron microscopy based techniques, transmission electron microscopy and atom probe tomography were performed revealing that minor variations in the heat treatment cause significant changes in
the resulting
microstructures (e.g. structural refinement, transition between diffusive and martensitic transformations). The experimental results on microstructure evolution during the applied different heat
treatment routes are discussed with respect to the mechanical properties.
Ductile Mg Alloys
Acta Materialia 60 (2012) 3011-3021
Acta Materialia vol 60 page 3011 ductile[...]
PDF-Dokument [1.0 MB]
The underlying mechanisms that are responsible for the improved room-temperature ductility in Mg–Y alloys compared to pure Mg are investigated by transmission electron microscopy and density functional theory. Both methods show a significant decrease in the intrinsic stacking fault I1 energy (I1 SFE) with the addition of Y. The influence of the SFE on the relative activation of different competing deformation mechanisms (basal, prismatic, pyramidal slip) is discussed. From this analysis we suggest a key mechanism which explains the transition from primary basal slip in hexagonal close-packed Mg to basal plus pyramidal slip in solid solution Mg–Y alloys. This mechanism is characterized by enhanced nucleation of hc + ai dislocations where the intrinsic stacking fault I1 (ISF1) acts as heterogeneous source for hc + ai dislocations. Possible electronic and geometric reasons for the modification of the SFE by substitutional Y atoms are identified and discussed.
Scientific REPOrtS | 7: 10458 | DOI:10.1038/s41598-017-10384-0
Scientific Reports 2017 ductile Magnesiu[...]
PDF-Dokument [2.8 MB]
Metals are the backbone of manufacturing owing to their strength and formability. Compared to polymers they have high mass density. There is, however, one exception: magnesium. It has a density of only 1.7 g/cm3, making it the lightest structural material, 4.5 times lighter than steels, 1.7 times lighter than aluminum, and even slightly lighter than carbon fibers. Yet, the widespread use of magnesium is hampered by its intrinsic brittleness. While other metallic alloys have multiple dislocation slip systems, enabling their well-known ductility, the hexagonal lattice of magnesium offers insufficient modes of deformation, rendering it intrinsically brittle. We have developed a quantum-mechanically derived treasure map which screens solid solution combinations with electronic bonding, structure and volume descriptors for similarity to the ductile magnesium-rare earth alloys. Using this insight we synthesized a surprisingly simple, compositionally lean, low-cost and industry-compatible new alloy which is over 4 times more ductile and 40% stronger than pure magnesium. The alloy contains 1 wt.% aluminum and 0.1 wt.% calcium, two inexpensive elements which are compatible with downstream recycling constraints.
Acta Materialia 57 (2009) 69–76
Acta Materialia 57 (2009) 69–76 ab initi[...]
PDF-Dokument [499.7 KB]
Ab initio calculations are becoming increasingly useful to engineers interested in designing new alloys, because these calculations are able to accurately predict basic material properties
only knowing the atomic composition of the material. In this paper, single crystal elastic constants of 11 bcc Mg–Li alloys are calculated using density functional theory (DFT) and compared with
available experimental
data. Based on DFT determined properties, engineering parameters such as the ratio of bulk modulus over shear modulus (B/G) and the ratio of Young’s modulus over mass density (Y/q) are
calculated. Analysis of B/G and Y/q shows that bcc Mg–Li alloys with 30–50 at.% Li offer the most potential as lightweight structural material. Compared with fcc Al–Li alloys, bcc Mg–Li alloys
have a lower B/G ratio, but a comparable Y/q ratio. An Ashby map containing Y/q vs B/G shows that it is not possible to increase both Y/q and B/G by changing only the composition of a
binary alloy.
Aluminium Solid Solution Alloys
Acta Materialia 85 (2015) 53-66
Acta Mater 85 (2015) 53 Ma et al - solid[...]
PDF-Dokument [2.3 MB]
We propose an approach for the computationally efficient and quantitatively accurate prediction of solid-solution strengthening. It combines the 2-D Peierls–Nabarro model and a recently developed solid-solution strengthening model. Solid-solution strengthening is examined with Al–Mg and Al–Li as representative alloy systems, demonstrating a good agreement between theory and experiments within the temperature range in which the dislocation motion is overdamped. Through a parametric study, two guideline maps of the misfit parameters against (i) the critical resolved shear stress, s0, at 0 K and (ii) the energy barrier, DEb, against dislocation motion in a solid solution with randomly distributed solute atoms are created. With these two guideline maps, s0 at finite temperatures is predicted for other Al binary systems, and compared with available experiments, achieving good agreement.