Ab initio based multiscale modeling of engineering materials
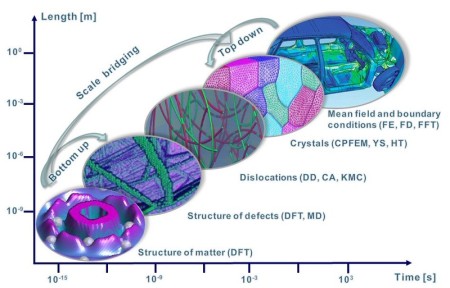

Materials science is based on identifying structure-property paradigms that are related to the material's hierarchical nature and yield a connection of macroscopic properties over multiple length and time scales. The lowest scale influencing these materials properties is governed by electrons being responsible for the chemical bonds between atoms. Many promising strategies of condensed matter theory are, therefore, based on the determination of the electronic structure (ES) of the considered materials and an intelligent transfer of its characteristics to higher-order scales using multidisciplinary schemes. More specifically, if the interaction of electrons is solely described using universal principles such as the fundamental laws of quantum mechanics condensed in the Schrödinger equation, these simulations are called first-principles, or ab initio methods.
It must be considered though that a solution of the quantum-mechanical Schrödinger equation including all relevant interactions between the electrons and atomic nuclei in solids is
computationally most demanding and hence only feasible for small systems. Consequently, practical ES calculations in solids were rather rare prior to the availability of high-speed computers. Even
with increasing computational resources, however, a series of approximations must be employed in order to render a comprehensive solution for any non-trivial system feasible. Despite these
limitations it is nowadays not only possible to mimic and actually predict certain experimental conditions and set-ups at an unprecedented level of accuracy without any experimental input, but also
to design completely new materials with tailored properties before actually synthesizing them.
One of the most established ab initio methods in materials science is the density functional theory (DFT) which has developed very rapidly in the last three
decades. It has now become a reliable and powerful tool for a vast spectrum of physical, chemical, metallurgical and engineering applications.
DFT is based upon Hohenberg-Kohn theorems which state that
i) all the physical properties of a system of interacting electrons are uniquely determined by its ground-state electron density distribution and
ii) there exists a universal energy functional that is minimized by the ground-state density.
As shown by Kohn and Sham, the problem of a system of interacting electrons can be then mapped onto an equivalent non-interacting problem. The ground-state charge-density distribution and the
non-interacting kinetic energy functional are given in terms of the auxiliary Kohn-Sham (KS) orbitals and the ground-state energy is expressed in terms of the KS eigenvalues. The KS orbitals are
found as solutions of non-linear (Schrödinger-like) equations, where potentials depend on their own eigenfunctions through the electron density distribution. The unknown functional of a system of
interacting electrons can be written as a sum of known ground-state kinetic energies of a system of non-interacting electrons, the classical electrostatic self-interaction of the electronic density,
and an unknown so-called exchange-correlation (xc) functional.
The exchange-correlation energy is the name given to the part of the energy functional that is not known and that must be approximated (e.g., local density approximation (LDA), generalized gradient approximation (GGA)). Once an explicit form for the xc energy is available, the KS equations can be solved in a self-consistent way using a variety of methods. When implemented, the KS orbitals are often expanded into a series of suitable basis functions, localized (such as, for example, atomic orbitals) in case of molecular systems, delocalized (e.g., plane waves) in case of periodic infinite crystals, or their combination. For more information see, e.g. The predictive strength of DFT-based computational approaches can be either used directly to determine scale-independent materials properties (such as the atomic density, Young's modulus, crystal structure, thermal expansion coefficient), or as an input for the up-scale transfer of materials parameters in multi-scale methods.
Some of the papers below by provide an introduction to this field.
Ab initio-guided design of twinning-induced plasticity steels:
page 320 MRS BULLETIN • VOLUME 41 • APRIL 2016
www.mrs.org/bulletin
MRS BULLETIN VOL 41 APRIL 2016 page 320 [...]
PDF-Dokument [946.9 KB]
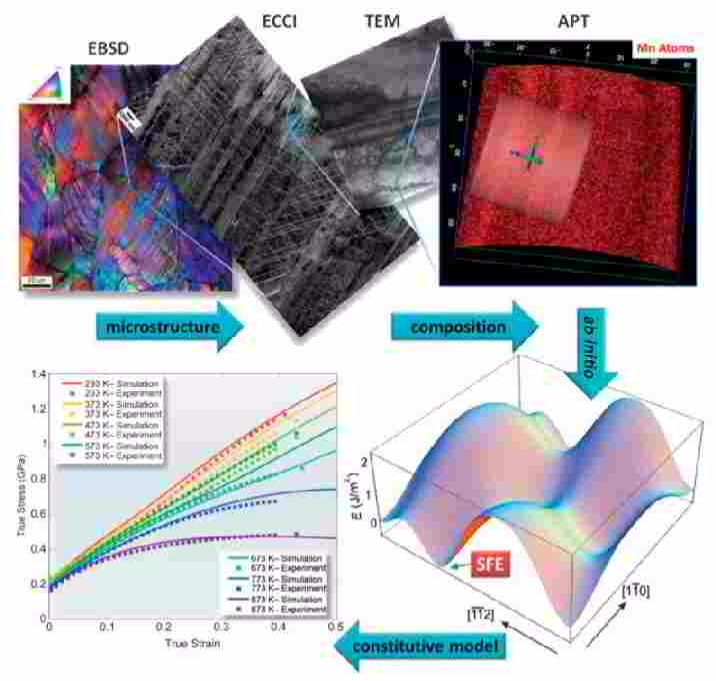 Ab initio guided alloy design of advanced steels: here - Fe-Mn TWIP steel
Ab initio guided alloy design of advanced steels: here - Fe-Mn TWIP steel
The twinning-induced plasticity effect enables designing austenitic Fe-Mn-C-based steels with >70% elongation with an ultimate tensile strength >1 GPa. These steels are characterized by high strain hardening due to the formation of twins and complex dislocation substructures that dynamically reduce the dislocation mean free path. Both mechanisms are governed by the stacking-fault energy (SFE) that depends on composition. This connection etween composition and substructure renders these steels ideal model materials for theory-based alloy design: Ab initio -guided composition adjustment is used to tune the SFE, and thus, the strain-hardening behavior for promoting the onset of twinning at intermediate deformation levels where the strain-hardening capacity provided by the dislocation substructure is exhausted. We present thermodynamic simulations and their use in constitutive models, as well as electron microscopy and combinatorial methods that enable validation of the strain-hardening mechanisms.
Duancheng Ma, Martin Friák, Johann von Pezold, Jörg Neugebauer, Dierk Raabe
Acta Materialia 98 (2015) 367-376
Ab initio study of compositional trends in solid solution strengthening in metals with low Peierls stresses
Acta Materialia 2015 ab initio solution [...]
PDF-Dokument [298.4 KB]
We identify and analyze general trends governing solid solution strengthening in binary alloys containing solutes across the Periodic table using quantum-mechanical calculations. Here we
present calculations for the model system of Al binary solid solutions. The identified trends originate from an approximately
parabolic dependence of two strengthening parameters to quantitatively predict the solid solution strengthening effect, i.e. the volume and slip misfit parameters. The volume misfit parameter
shows a minimum (concave-up behavior) as a function of the solute element group number in the periodic table, whereas the slip misfit parameter shows a maximum (concave-down behavior). By
analyzing reported data, a similar trend is also found in Ni and Mg (basal slip) binary systems. Hence, these two strengthening parameters are strongly anti-correlated, which can be understood
in terms of the Fermi level shift in
the framework of free electron model. The chemical trends identified in this study enable a rapid and efficient identification of the solutes that provide optimum solid–solution strengthening.
The approach described here may thus serve as basis for ab initio guided metallurgical materials design.
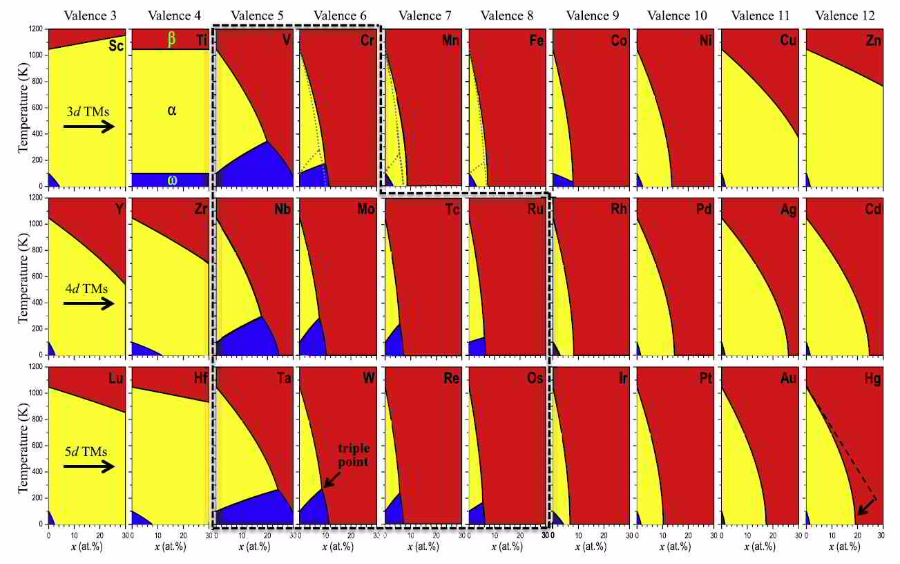 Metastable phase diagrams of Tietransition metal (TM) alloys computed from ab initio.
Metastable phase diagrams of Tietransition metal (TM) alloys computed from ab initio.
Acta Materialia 113 (2016) 311-319
Acta Materialia 113 (2016) 311 ab initio[...]
PDF-Dokument [2.5 MB]
Acta Materialia 85 (2015) 53-66
Acta Materialia 2015 D Ma et al solid so[...]
PDF-Dokument [1.1 MB]
We propose an approach for the computationally efficient and quantitatively accurate prediction of solid-solution strengthening. It combines the 2-D Peierls–Nabarro model and a recently developed solid-solution strengthening model. Solid-solution strengthening is examined with Al–Mg and Al–Li as representative alloy systems, demonstrating a good agreement between theory and experiments within the temperature range in which the dislocation motion is overdamped. Through a parametric study, two guideline maps of the misfit parameters against (i) the critical resolved shear stress, s0, at 0 K and (ii) the energy barrier, DEb, against dislocation motion in a solid solution with randomly distributed solute atoms are created. With these two guideline maps, s0 at finite temperatures is predicted for other Al binary systems, and compared with available experiments, achieving good agreement.
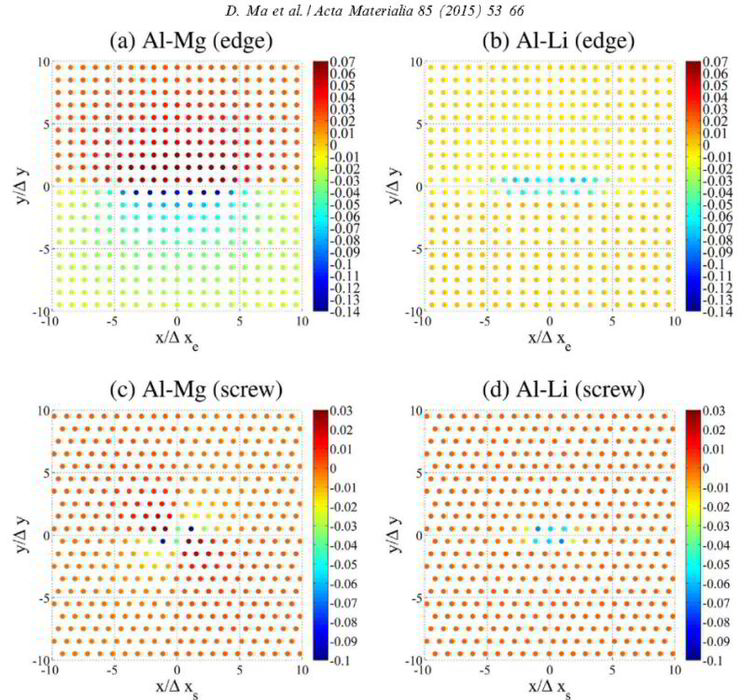 Ma, Acta Mater 85 (2015) 53: Position-dependent dislocation–solute interaction energy in Al–Mg (a and c) and Al–Li (b and d).
Ma, Acta Mater 85 (2015) 53: Position-dependent dislocation–solute interaction energy in Al–Mg (a and c) and Al–Li (b and d).
Acta Materialia 76 (2014) 94-105
Acta Mater 2014 Fe–Mn maraging steels wi[...]
PDF-Dokument [2.6 MB]
B2 NiMn and Ni2MnAl Heusler nanoprecipitates are designed via elastic misfit stabilization in Fe–Mn maraging steels by combining transmission electron microscopy (TEM) correlated atom probe
tomography (APT) with ab initio simulations. Guided by these predictions, the Al content of the alloys is systematically varied, and the influence of the Al concentration on structure stability, size
and distribution of precipitates formed during ageing at 450 °C is studied using scanning electron microscopy–electron backscatter diffraction,
TEM and APT. Specifically, the Ni2MnAl Heusler nanoprecipitates exhibit the finest sizes and highest dispersion and hence lead to significant strengthening. The formation of the different types of
precipitates and their structure, size, dispersion and effect on the mechanical properties of the alloys are discussed.
 Acta Materialia 76 (2014) 94: Heusler nano-precipitates in maraging steel
Acta Materialia 76 (2014) 94: Heusler nano-precipitates in maraging steel
The I1 intrinsic stacking fault energy (I1 SFE) serves as an alloy design parameter for ductilizing Mg alloys. In view of this effect we have conducted quantum–mechanical calculations for Mg15X soli
Ductility of Mg rare earth alloys ab ini[...]
PDF-Dokument [4.6 MB]
Acta Materialia 75 (2014) 147–155
Acta Materialia 75 (2014) 147 nanodiffus[...]
PDF-Dokument [787.4 KB]
A key requirement of modern steels – the combination of high strength and high deformability – can best be achieved by enabling a local adaptation of the microstructure during deformation. A local hardening is most efficiently obtained by a modification of the stacking sequence of atomic layers, resulting in the formation of twins or martensite. Combining ab initio calculations with in situ transmission electron microscopy, we show that the ability of a material to incorporate such stacking faults depends on its overall chemical composition and, importantly, the local composition near the defect, which is controlled by nanodiffusion. Specifically, the role of carbon for the stacking fault energy in high-Mn steels is investigated. Consequences for the long-term mechanical properties and the characterisation of these materials are discussed.
Multi-methodological approaches combining quantum-mechanical and/or atomistic simulations with continuum methods have become increasingly important when addressing multi-scale phenomena in computation
overview-combining ab initio with contin[...]
PDF-Dokument [1.5 MB]
Solid-solution strengthening in six Al–X binary systems is investigated using first-principle methods. The volumetric mismatch parameter and the solubility enthalpy per solute were calculated. We de
ab-initio-solid-solution.pdf
PDF-Dokument [471.3 KB]
Acta Materialia 55 (2007) 4475-4487
Acta Materialia 55 (2007) 4475–4487 ab i[...]
PDF-Dokument [1.9 MB]
In this study we present a new strategy for the theory-guided bottom up design of beta-Ti alloys for biomedical applications using a quantum mechanical
approach in conjunction with experiments. Parameter-free density functional theory calculations are used to provide theoretical guidance in selecting and optimizing Ti-based
alloys with respect to three constraints: (i) the use of non-toxic alloy elements; (ii) the stabilization of the body centered cubic b-phase at room temperature; (iii) the reduction of the
elastic stiffness compared to
existing Ti-based alloys. Following the theoretical predictions, the alloys of interest are cast and characterized with respect to their crystallographic
structure, microstructure, texture, and elastic stiffness. Due to the complexity of the ab initio calculations, the
simulations have been focused on a set of binary systems of Ti with two different high melting body-centered cubic metals, namely, Nb and Mo. Various levels of model approximations to
describe mechanical and thermodynamic properties are tested and critically evaluated.
The experiments are conducted both, on some of the binary alloys and on two more complex engineering alloy variants, namely, Ti–35 wt.% Nb–7 wt.% Zr–5 wt.% Ta and Ti–20 wt.% Mo–7 wt.% Zr–5 wt.%
Ta.
 Jointly using ab initio and FEM approaches to design hip implants (Acta Materialia 55 (2007) 4475)
Jointly using ab initio and FEM approaches to design hip implants (Acta Materialia 55 (2007) 4475)
Acta Materialia 57 (2009) 69: Ab initio calculations are becoming increasingly useful to engineers interested in designing new alloys, because these calculations are able to accurately predict basic material properties only knowing the atomic composition of the material. In this paper, single crystal elastic constants of 11 bcc Mg–Li alloys are calculated using density functional theory (DFT) and compared with available experimental data. Based on DFT determined properties, engineering parameters such as the ratio of bulk modulus over shear modulus (B/G) and the ratio of Young’s modulus over mass density (Y/q) are calculated. Analysis of B/G and Y/q shows that bcc Mg–Li alloys with 30–50 at.% Li offer the most potential as lightweight structural material. Compared with fcc Al–Li
Acta Materialia 57 (2009) 69–76 ab initi[...]
PDF-Dokument [499.7 KB]
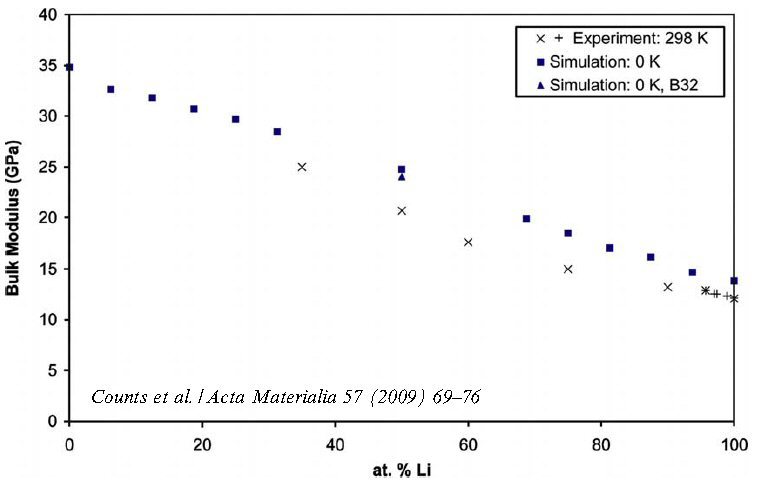
Acta Materialia 100 (2015) 90-97
Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one
Duancheng Ma, Blazej Grabowski, Fritz Körmann, Jörg Neugebauer, Dierk Raabe
Acta Mater 100 (2015) 90 Ab initio therm[...]
PDF-Dokument [1.1 MB]
High entropy alloy (HEA) refers to a multi-component (5 or more) single-phase solid-solution alloy in or near an equi-atomic composition [1–3]. The basic idea behind HEAs is an exploration of
phase diagrams which are not confined to regions of a single major base element as in conventional alloy design. The aim of the new concept is a development of new alloy systems with novel mechanical
and functional properties [1–4]. The term ‘‘high entropy” refers to the high configurational entropy which is deemed to be responsible for the stability of the single solid solution phase over
competing intermetallic and elemental phases
[1,2]. The configurational entropy is however only one out of various contributions to the Gibbs free energy and it is the latter that
determines phase stabilities. It is therefore of crucial importance to also investigate the role of (1) the formation enthalpy and (2) other entropy contributions.
The role of the formation enthalpy has been recently addressed by [5]. The authors showed that among various multi-component alloys with equal ideal configurational entropy only one was sufficiently
stabilized to form a single-phase solid-solution. In all other multi-component alloys studied in Ref. [5], the formation enthalpy was dominant over the configurational entropy resulting in the
formation of intermetallic phases. The impact of entropy contributions other than the configurational one on the phase stability of HEAs has so far only scarcely been addressed. Rather, with
reference to the first core effect, it has been implicitly assumed that entropic contributions related to atomic vibrations, electronic and magnetic excitations are negligible in determining
phase
stabilities in HEAs.
In the present paper we employ ab initio based simulations to investigate the validity of the latter assumption for the prototype HEA CoCrFeMnNi [6–10]. In particular, we study the phase stability
between hcp, fcc, and bcc structures by calculating the free energies of the respective crystal structures at T = 0 K as well as at finite temperatures, including a detailed analysis of the balance
between the different entropy contributions (vibrational, electronic, magnetic
as well as configurational).
More specific we investigate the thermodynamic properties of the prototype equi-atomic high entropy alloy (HEA) CoCrFeMnNi by using finite-temperature ab initio methods. All relevant free energy
contributions are considered for the hcp, fcc, and bcc structures, including electronic, vibrational, and magnetic excitations. We predict the paramagnetic fcc phase to be most stable above room
temperature in agreement with experiment. The corresponding thermal expansion and bulk modulus agree likewise well with experimental measurements. A careful analysis of the underlying entropy
contributions allows us to identify that the originally postulated dominance of the configurational entropy is questionable. We show that vibrational, electronic, and magnetic entropy contributions
must be considered on an equal footing to reliably predict phase stabilities in HEA systems.
Acta Materialia vol 60 (2012) pages 3011-3021
The relation between ductility and stacking fault energies in Mg and Mg–Y alloys
S. Sandlöbes, M. Friak, S. Zaefferer, A. Dick, S. Yi, D. Letzig, Z. Pei, L.-F. Zhu, J. Neugebauer, D. Raabe
Acta Materialia 60 (2012) 3011 ductility[...]
PDF-Dokument [1.0 MB]
 Acta Materialia vol 60 (2012) pages 3011-3021 The relation between ductility and stacking fault energies in Mg and Mg–Y alloys S. Sandlöbes, M. Friak, S. Zaefferer, A. Dick, S. Yi, D. Letzig, Z. Pei, L.-F. Zhu, J. Neugebauer, D. Raabe
Acta Materialia vol 60 (2012) pages 3011-3021 The relation between ductility and stacking fault energies in Mg and Mg–Y alloys S. Sandlöbes, M. Friak, S. Zaefferer, A. Dick, S. Yi, D. Letzig, Z. Pei, L.-F. Zhu, J. Neugebauer, D. Raabe
 Acta Materialia vol 60 (2012) pages 3011-3021 The relation between ductility and stacking fault energies in Mg and Mg–Y alloys S. Sandlöbes, M. Friak, S. Zaefferer, A. Dick, S. Yi, D. Letzig, Z. Pei, L.-F. Zhu, J. Neugebauer, D. Raabe
Acta Materialia vol 60 (2012) pages 3011-3021 The relation between ductility and stacking fault energies in Mg and Mg–Y alloys S. Sandlöbes, M. Friak, S. Zaefferer, A. Dick, S. Yi, D. Letzig, Z. Pei, L.-F. Zhu, J. Neugebauer, D. Raabe
Magnesium and its alloys are the lightest engineering metallic materials. They have superior potential in the fields of ultra-lightweight mobility and energy-related applications
(e.g. [1,2]). While pure Mg and most commercial wrought magnesium alloys exhibit a low room-temperature ductility (about 5% elongation for pure Mg) the addition of rare earth
elements in solid solution causes a significant increase in ductility (e.g. [3–6]): Mg and single-phase solid-solution
Mg–Y alloys showed an increase in room-temperature ductility by about 5 times, while maintaining comparable strength and well-balanced work hardening, through the addition of 3 wt.% Y
[7]. In order to obtain deeper insight into the fundamental mechanisms responsible for this solid-solution driven ductility enhancement, density functional theory (DFT)-based methods
provide a new tool to quantitatively address possible effects of Y on plasticity in these alloys [8–11]. Understanding the electronic, magnetic and geometric effects of solutes on
crystalline metals at the atomic scale opens a pathway from our current phenomenological descriptive picture towards a more physics-based
predictive understanding of the thermodynamic and kinetic properties of metallic materials (e.g. [8–12]). Such an approach is particularly feasible when minor alloy changes (such as in
the case of solid solution Mg–Y) lead to substantial improvements in the macroscopic behaviour. Following this approach, we present here a complementary study via joint DFT- and
transmission electron
microscopy (TEM)-based methods to understand the structural atomistic and electronic influence of Y on the mechanical behaviour of Mg alloys.
Here, the underlying mechanisms that are responsible for the improved room-temperature ductility in Mg–Y alloys compared to pure Mg are investigated by transmission electron microscopy and density functional theory. Both methods show a significant decrease in the intrinsic stacking fault I1 energy (I1 SFE) with the addition of Y. The influence of the SFE on the relative activation of different competing deformation mechanisms (basal, prismatic, pyramidal slip) is discussed. From this analysis we suggest a key mechanism which explains the transition from primary basal slip in hexagonal close-packed Mg to basal plus pyramidal slip in solid solution Mg–Y alloys. This mechanism is characterized by enhanced nucleation of hc + ai dislocations where the intrinsic stacking fault I1 (ISF1) acts as heterogeneous source for hc + ai dislocations. Possible electronic and geometric reasons for the modification of the SFE by substitutional Y
atoms are identified and discussed.
Elastic properties of face-centred cubic Fe–Mn–C studied by
nanoindentation and ab initio calculations
S. Reeh, D. Music, T. Gebhardt, M. Kasprzak, T. Jäpel, S. Zaefferer, D. Raabe, S. Richter, A. Schwedt, J. Mayer, B. Wietbrock, G. Hirt, J.M. Schneider
Acta Materialia 60 (2012) 6025-6032
Reeh_AM_2012-ab-initio-elastic-modul.pdf
PDF-Dokument [887.1 KB]
 Elastic properties of face-centred cubic Fe–Mn–C studied by nanoindentation and ab initio calculations S. Reeh, D. Music, T. Gebhardt, M. Kasprzak, T. Jäpel, S. Zaefferer, D. Raabe, S. Richter, A. Schwedt, J. Mayer, B. Wietbrock, et al.
Elastic properties of face-centred cubic Fe–Mn–C studied by nanoindentation and ab initio calculations S. Reeh, D. Music, T. Gebhardt, M. Kasprzak, T. Jäpel, S. Zaefferer, D. Raabe, S. Richter, A. Schwedt, J. Mayer, B. Wietbrock, et al.
Mn-rich transformation-induced plasticity/twinning-induced plasticity (TRIP/TWIP) steels reveal outstanding mechanical properties combining high strength (>1000 MPa) and superior
ductility (elongation to failure of >50%) [1,2]. These exceptional properties are due to dislocation glide, mechanical twinning and strain-induced epsilon- and alpha-martensitic
transformations [1]. The formation of twins (TWIP effect) or multiple martensitic transformations (TRIP effect) under mechanical load is affected by the stacking fault energy (SFE),
which is dependent on temperature and chemical composition [3,4]. The influence of SFE [4], temperature [4,5] and strain rate [5] on the deformation
mechanisms and the interactions among the various deformation mechanisms of face-centred cubic (fcc) Fe–Mn–C steels has been studied in a number of papers [6,7]. In contrast the
C-induced changes on the elastic properties of high Mn steels have not been studied systematically before.
For the binary fcc Fe–Mn random alloys elastic properties have been reported by Music et al. [8], Gebhardt et al. [9] and Cankurtaran et al. [10]. Even though these alloys are
antiferromagnetic (AFM) with Ne´el temperatures well above room temperature, their elastic properties are well predicted using the disordered local moment (DLM) approach [8,9] to
describe the magnetic configuration.
With magnetically ordered configurations, such as AFM, the stiffness of these random alloys is overestimated due to lack of lattice softening [9]. However, there are no systematic data
on the dependence of the Young’s modulus
values on the C concentration in ternary fcc Fe–Mn–C alloys.
In this paper, the influence of the C and Mn content on the Young’s modulus of fcc Fe–Mn–C steel thin film synthesized by combinatorial sputter deposition is
systematically investigated and compared to the same data obtained from bulk samples. Ab initio calculations are carried out to determine the Young’s modulus of selected Fe–Mn–C alloys
and to correlate these with the electronic structure.
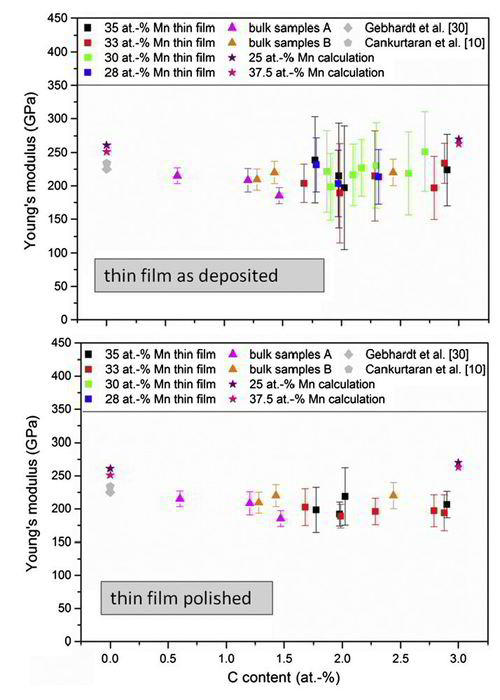
More specific we have studied experimentally and theoretically the influence of C and Mn content on the Young’s modulus of Fe–Mn–C alloys. Combinatorial thin film and bulk samples were characterized regarding their structure, texture and Young’s modulus. The following chemical composition range was investigated: 1.5–3.0 at.% C, 28.0–37.5 at.% Mn and 60.6–69.8 at.% Fe. The experimental lattice parameters change marginally within 3.597–3.614A ˚ with the addition of C and are consistent with ab initio calculations. The Young’s modulus data are in the range of 185 ± 12–251 ± 59 GPa for the bulk samples and the thin film, respectively. C has no significant effect on the Young’s modulus of these alloys within the composition range studied here. The ab initio calculations are 15–22% larger than the average Young’s modulus values of the as-deposited and polished thin film at 3 at.% C. The comparison of thin film and bulk samples results reveals similar elastic properties for equivalent compositions, indicating that the applied research strategy consisting of the combinatorial thin film approach in conjunction with ab initio calculations is useful to study the composition dependence of the structure and elastic properties of Fe–Mn–C alloys. The very good agreement between the presented calculations and the experimentally determined lattice parameters and Young’s modulus values implies that the here-adopted simulation strategy yields a reliable description of carbon in Fe–Mn alloys, important for future alloy design.
materials-05-01853-v2.pdf
PDF-Dokument [722.5 KB]
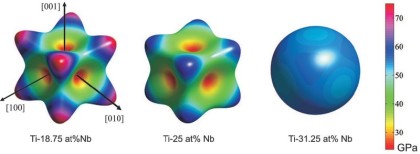
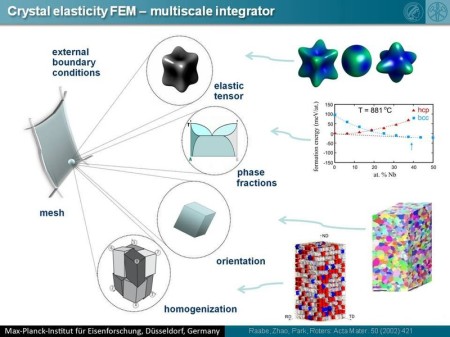
A necessary prerequisite for a successful theory-guided up-scale design of materials with application-driven elastic properties is the availability of reliable homogenization techniques. We report on
Hajjir-MRS-proceedings-2012.pdf
PDF-Dokument [275.4 KB]
Ab Initio Guided Design of bcc Ternary Mg–Li–X (X¼Ca, Al, Si, Zn, Cu) Alloys for Ultra-Lightweight Applications
William Art Counts, Martin Friak, Dierk Raabe and Jörg Neugebauer
ADVANCED ENGINEERING MATERIALS 2010, 12, No. 7, page 572
ADVANCED ENGINEERING MATERIALS 2010 vol [...]
PDF-Dokument [412.8 KB]
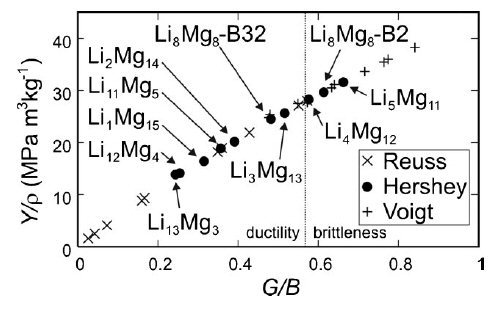 Ashby map of Y/r versus G/B of bcc Mg–Li alloys. The vertical dotted line separates the ductile and brittle regions.[15] The map nicely shows that it is not possible to increase Y/r without increasing G/B (i.e., tendency toward the brittleness).
Ashby map of Y/r versus G/B of bcc Mg–Li alloys. The vertical dotted line separates the ductile and brittle regions.[15] The map nicely shows that it is not possible to increase Y/r without increasing G/B (i.e., tendency toward the brittleness).
 Ashby map of Y/r versus G/B for bcc MgLi-based ternaries with the master curve of Mg–Li binary system (third-order polynomial fit, solid line) where every data point is accompanied with the name of ternary solute X (Ca, Al, Si, Cu, Zn) in Mg-Li-X alloys,
Ashby map of Y/r versus G/B for bcc MgLi-based ternaries with the master curve of Mg–Li binary system (third-order polynomial fit, solid line) where every data point is accompanied with the name of ternary solute X (Ca, Al, Si, Cu, Zn) in Mg-Li-X alloys,
The global challenges regarding energy savings and CO2 reduction with the current worldwide trend toward an ever-increasing demand for global mobility are among the most essential tasks in
materials science and engineering. Any successful attempt to achieve both goals requires the development of novel ultra-lightweight metallic materials with excellent mechanical
and functional properties. This applies for both fossile-fuel and battery-driven units. In this context novel Mg alloys play a key role owing to their low mass
density.[1–5] Pure Mg is a common, lightweight metal that can potentially be more widely applied as a structural material but the use of wrought Mg- and
Mg-alloys is limited in large-scale manufacturing operations: Mg and Mg-alloys are difficult to deform at room temperature, and once formed they often
reveal undesirable mechanical properties. In this article we address two important aspects related to Mg alloys. First, how can the forming and anisotropy properties
of Mg alloys be improved? These properties are of particular relevance for instance in the field of advanced automotive manufacturing. Second, can we employ new alloy
design strategies to achieve a more knowledge-driven rapid maturation of metallic materials that go beyond conventional empirical design approaches?
Regarding the first issue, we aim in this work at designing Mg alloys crystallizing in a phase that is more easily formable. The lack of room temperature ductility in Mg alloys is primarily due to
the near-hexagonal closed packed (hcp) crystal structure of Mg. In hcp Mg, the critical resolved shear stress of the two basal slip systems is much lower than that
of other slip systems. The two available slip systems are not enough to fulfill the von Mises criterion that requires five independent slip systems to accommodate an arbitrary deformation state via
dislocation glide. The absence of room temperature ductility and the evolution of strong deformation textures are inherent limitations of the hcp crystal structure that to date have not been
completely overcome.[6–10] Changing the crystal structure fully or partially from hcp to
either face centered cubic (fcc) or body centered cubic (bcc) is one way to realize Mg alloys that are less anisotropic and mechanically more compliant as this change increases the number of
plastic degrees of freedom (higher number of available slip systems). The consequence would be better plastic formability at room temperature and a reduced tendency to form
disadvantageous deformation textures. Cubic Mg would therefore be very attractive from a manufacturing point of view. A number of Mg–X alloys are known to have a cubic structure. For
example, the Mg–Li system has a stable bcc phase at room temperature and the bcc phase is stabilized with as little as 30 at% Li. Li does not only stabilize the bcc crystal
structure; it also has the advantage of further decreasing the density of the Mg alloy since Li is the lightest known metal. The resulting Mg-Li alloys are therefore
among the lightest possible metallic alloys. Li additions on the other hand reduce the overall stiffness of Mg–Li alloys. Therefore, a
systematic materials-design search (e.g., see Refs.[11–14]) is needed that accomplishes an optimization with respect to multiple, in part even contradicting, materials criteria. This leads us to
the second main point of how such a more systematic and knowledge-driven optimization can be pursued. In this context we choose a quantum mechanics-based bottom-up approach to
identify more rapidly not only suited compositions with regard to the thermodynamic stabilization of the desired phase but also scrutinize some of the basic structural and mechanical features of
possible alloy candidates. In contrast to many top-down approaches that start from the macroscopic scale and continue downscale, here we are starting at the most fundamental level of materials
science – the quantum-mechanical level – which describes the interaction of the fundamental constituents of any material without any experimental input. The only parameters entering the
quantum-mechanical calculations are the nucleus number of the specific chemical element. These methods are called first principles methods or, using the corresponding Latin term, ab initio
methods. In order to carry out an ab initio guided materials-design strategy, a multi-disciplinary approach has been used in this paper. First, ground-state density functional
theory (DFT) calculations were performed for a set of bcc and hcp ordered alloys in order to obtain information on thermodynamic phase-stabilities and linear-elasticity data at the
atomic level.
Second, polycrystalline elastic moduli and other engineering parameters measurable at macroscale were predicted employing linear-elasticity homogenization techniques that allow bridging the
scale difference between atomistic and macroscopic levels. Using this methodological strategy, an alloy composition with desired properties may be suggested employing the following iterative
steps. First, starting from an initial composition its properties are computed and compared to desired ones. Based on the residuum/deviation
of the properties on the macroscale a new atomic composition is suggested and studied. These cycles are repeated until the desired properties are obtained. Of course, if the properties are not
accessible by any chemical composition, a new set of properties has to be identified.In this paper, we start with the Mg–Li bcc binary system. It was shown that (i) the resulting materials properties
compare very well with available experimental data, (ii) polycrystalline elastic constants (Young’s modulus Y and shear modulus G) possess nonlinear variations as a function of alloy
composition, (iii) alloys with about 30 at% Li were the stiffest, and (iv) alloys with greater than 70 at% Li were the softest.[13,14] Using
the DFT calculated data, an Ashby map containing (i) Young’s specific modulus Y/r as a measure of the strength and (ii) the B/G as a suggested indicator of either brittle or ductile behavior was
previously constructed.[15] Plotting the Reuss, Voigt, and self-consistent Hershey values of various local arrangements together in this map resulted in a universal master curve dependence that
links increasing Y/r with decreasing B/G, i.e., increasing tendency toward an inherent brittleness. That such a universal master curve exists indicates that there is a fundamental
materials-design limitation, i.e., it is not possible to increase both Y/r and B/G by changing only the composition or local order of a binary alloy. Here we show that the product of Y/r and G/B
(i.e., an inverse of the originally proposed B/G ratio) is actually nearly constant for all studied bcc Mg–Li compounds. Next, in an attempt to bypass the identified materials-design limitation,
the stoichiometric MgLi was chosen as a test system in which a ternary alloying was studied employing ab initio calculated thermodynamic and elasticity information. Specifically, properties of a set
of Mg-Li–X ternaries (X¼Ca, Al, Si, Cu, Zn) were determined. It is shown that the studied solute additions have rather detrimental impact on the pair of studied materials properties and are not able
to overcome the limitation. Thus in summary we show here that ab initio calculations are becoming increasingly important for designing new alloys as these calculations can accurately predict basic
structural, mechanical, and functional properties using only the atomic composition as a basis. In this paper, fundamental physical properties (like formation energies and elastic constants) of a set
of bcc Mg–Li and Mg–Li-based compounds are calculated using density functional theory (DFT). These DFT-determined properties are in turn used to calculate engineering parameters such
as (i) specific Young’s modulus (Y/r) or (ii) shear over bulk modulus ratio (G/B) differentiating between brittle and ductile behavior. These parameters are then used to identify those alloys that
have optimal mechanical properties for lightweight structural applications. First, in case of the binary Mg–Li system, an Ashby map containing Y/r versus G/B shows that it is not possible to increase
Y/r without simultaneously increasing G/B (i.e., brittleness) by changing only the composition of a binary alloy. In an attempt to bypass such a fundamental materials-design limitation, a set of
Mg–Li–X ternaries (X¼Ca, Al, Si, Cu, Zn) based on stoichiometric Mg–Li with CsCl structure was studied. It is shown that none of the studied ternary solutes is able to simultaneously improve
both specific Young’s modulus and ductility.
Hence thsi work is about new approaches to the identification of fundamental materials-design limits in ultra
light-weight Mg-Li alloys via quantum-mechanical calculations.