Combinatoral Bulk Metallurgical Alloy Design
Why do we need combinatorial metallurgical alloy design?
Today, after more than 4000 years of development, we still use only about 1000 different types of metallic alloys out of a sheer infinite combinatorial space of about 1060 possible combinations when considering only the 50-60 frequently used elements. This means that we only stand at the beginning of basic research of metals.
Yet, on the other hand, increased efficiency in the development of novel metallic structural materials is a key to providing fundamental and applied alloy solutions in the fields of energy, mobility, health, infrastructure and safety. Examples of the associated basic metallurgical questions are alloy- and processing- sensitive changes in complex strain hardening phenomena. In high strength steels these are often associated with displacive transformations, such as twinning and transformation-induced plasticity (TWIP and TRIP), which can be tuned through variations in the stacking fault energy and microstructure. Associated engineering issues are systematic analysis of the corresponding trends in texture evolution, sheet forming and damage parameters, joining behaviour, hydrogen susceptibility or fatigue. In thin film systems, in which combinatorial high throughput methods were invented and established, substantial progress has been made in a number of materials design fields, such as shape memory alloys, MAX phases and shape change materials.
What are the differences between sputtered thin film and bulk combinatorial alloy design methods ?
Such combinatorial methods have mostly been applied to the design of functional materials, where, typically, the intrinsic composition-dependent properties dominate over the relevance of the microstructure. In structural alloys, on the other hand, the relevant length scales that determine the mechanical behaviour are parameters such as grain size, texture, precipitate dispersion and topology and the dislocation cell structure, to name but a few. The correlation lengths associated with such lattice defects are usually of the order of several nanometres to multiple micrometres, hence, they usually exceed the dimensions accessible in thin films. This means that the mechanical properties of structural materials are not only determined by their chemical composition, but to a large extent by the microstructure, which in turn is greatly influenced by the thermo-mechanical treatments (TMTs) applied after synthesis. Consequently, the current conventional approach for the experimentally guided development of metallic structural materials typically consists of a number of iterative loops that include the following basic steps: Bulk casting of a single composition charge, hot and/or cold deformation (e.g. conventional or thermo-mechanical rolling), heat treatment (such as quenching, partitioning or ageing), machining of tensile specimens and mechanical testing.
How can large-scale bulk combinatorial metallurgical screening of new steels be realized ?
Coupled with the wide range of possible chemical compositions and different TMT setups, this kind of incremental trial and error approach typically provides robust mechanical data on novel alloys, however, it is often too time consuming for a more efficient investigation and the maturation of complex alloy systems as a function of composition, TMT processing and microstructure.
Thus it becomes clear that the development of novel and more efficient high throughput methods for the bulk synthesis, TMT processing and following investigations of structural materials is of
great interest. It is aimed at drastically
increasing both the scientific and engineering efficiency by reducing the time between an alloy design idea and final evaluation of the material’s mechanical and microstructural properties from
several weeks or even months down to hours. Materials exhibiting desirable properties (“hits”) can be readily selected from the investigated composition/TMT matrix and then scaled up via
conventional methods for more detailed investigations based on the information collected. Rapid and systematic screening of prototype alloys and TMT setups, ideally employed in concert with
theory-based, alloy-sensitive simulation
methods, thereby allows the efficient and knowledge-based design of structural materials. Steels containing high amounts of manganese (Mn), aluminium (Al) and carbon (C) were here chosen as an
example material system to utilize the novel approach outlined above. Also referred to as “Triplex” steels, these materials exhibit a low mass
density, high strength and excellent ductility
in comparison with established steels for structural engineering applications. The authors derived this attractive property profile from an austenitic or austenitic/ ferritic matrix, both stabilized
and strengthened by alloying iron (Fe) with Mn (18–28 wt.%) and C (0.7–1.2 wt.%), together with Al addition (3–12 wt.%) for low specific weight and improved corrosion resistance.
Profound changes in the mechanical properties can be achieved by ageing the material after solution annealing and quenching. More specifically, kappa-Al(Fe,Mn)3C carbides were found to precipitate from the matrix during ageing, growing from C-enriched areas, most probably formed via spinodal decomposition during quenching. The
further development of Fe–Mn–Al–C alloys must involve simultaneous investigation of the respective microstructural phenomena by high resolution characterisation techniques such as atom probe
tomography (APT) and transmission electron microscopy (TEM). Such techniques, however, require increased efforts in sample preparation and experimental procedures and are, therefore, preferably
applied only to those compositions and microstructures that reveal the most interesting characteristics or properties. The wide range of possible chemical compositions in the Fe–Mn–Al–C quaternary system, combined with a large matrix of possible TMT routes (especially ageing time and temperature), justifies the high level
of motivation for the development and use of rapid alloy prototyping (RAP) techniques as an efficient tool for structural materials development.
Hence, the aim of this webpage is to present several new approaches to the rapid investigation of bulk metallic structural materials, whereby the mechanical properties of a group of alloys
systematically varied in terms of chemical
composition and the imposed TMT treatments can be evaluated simultaneously and thus with a higher throughput than with conventional methods and step by step iteration of these parameters. The
approach is referred to as RAP.
As an exemple here we also present - among others - a novel group of 30Mn–1.2C steels in terms of the effects of varying Al content and different ageing conditions on the microstructure and
properties. Selected results are directly compared with data obtained from conventionally synthesised and processed samples to validate the approach.
Development of high-entropy alloys by combinatorial metallurgical synthesis and processing
High-entropy alloys (HEAs) with multiple principal elements open up a practically infinite space for
designing novel materials. Probing this huge material universe requires the use of combinatorial and high-throughput synthesis and processing methods. Here, we present and discuss four
different combinatorial experimental methods that have been used to accelerate the development of novel HEAs, namely, rapid alloy prototyping,
diffusion-multiples, laser additive manufacturing, and combinatorial co-deposition of thin-film materials libraries. While the first
three approaches are bulk methods which allow for downstream processing and microstructure adaptation, the latter technique is a thin-film method capable of efficiently synthesizing
wider ranges of composition and using high-throughput measurement techniques to characterize their structure and properties. Additional coupling of these high-throughput experimental
methodologies
with theoretical guidance regarding specific target features such as phase (meta)stability allows for effective screening of novel HEAs with beneficial property profiles.
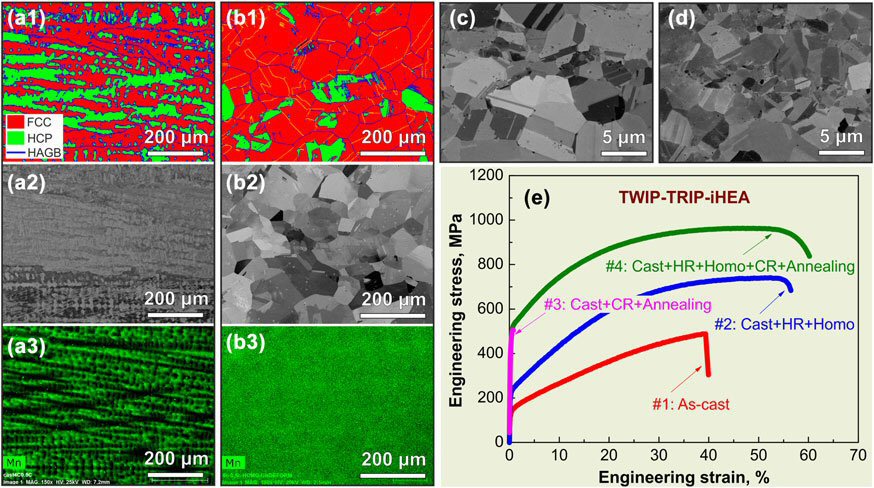 Microstructure, compositional homogeneity state, and tensile properties of an iHEA with nominal composition of Fe49.5Mn30Co10Cr10C0.5 (at.%) in various processing conditions obtained after specific steps of Metallurgical Combinatorial Design and Synthesis
Microstructure, compositional homogeneity state, and tensile properties of an iHEA with nominal composition of Fe49.5Mn30Co10Cr10C0.5 (at.%) in various processing conditions obtained after specific steps of Metallurgical Combinatorial Design and Synthesis
J. Mater. Res., 2018
REVIEW J. Mater. Res. 2018 Combinatorial[...]
PDF-Dokument [876.3 KB]
Nature Materials Reviews Aug 2019 High-e[...]
PDF-Dokument [2.4 MB]
Revealing the relationships between chemistry, topology and stiffness of ultrastrong Co-based metallic glass thin films: A combinatorial approach
Acta Materialia 107 (2016) 213-219
Acta Materialia 107 (2016) 213 combinato[...]
PDF-Dokument [2.3 MB]
An efficient way to study the relationship between chemical composition and mechanical properties of thin films is to utilize the combinatorial approach, where spatially resolved mechanical property measurements are conducted along a concentration gradient. However, for thin film glasses many properties including the mechanical response are affected by chemical topology. Here a novel method is introduced which enables spatially resolved short range order analysis along concentration gradients of combinatorially synthesized metallic glass thin films. For this purpose a CoZrTaB metallic glass film of 3 mm thickness is deposited on a polyimide foil, which is investigated by high energy X-ray diffraction in transmission mode. Through the correlative chemistry-topology-stiffness investigation, we observe that an increase in metalloid concentration from 26.4 to 32.7 at% and the associated formation of localized (hybridized) metal e metalloid bonds induce a 10% increase in stiffness. Concomitantly, along the same composition gradient, a metalloid-concentration-induced increase in first order metal - metal bond distances of 1% is observed, which infers itinerant (metallic) bond weakening. Hence, the metalloid concentration induced increase in hybridized bonding dominates the corresponding weakening of metallic bonds.
Synergy of atom-probe structural data and quantum-mechanical calculations in a theory-guided design of extreme-stiffness superlattices containing metastable phases
M. Friák,D. Tytko, D. Holec, P-P Choi, P. Eisenlohr, D. Raabe and J. Neugebauer
New J Physics 17 (2015) 093004 extreme s[...]
PDF-Dokument [1.5 MB]
A theory-guidedmaterials design of nano-scaled superlattices containing metastable phases is critically important for future development of advanced lamellar compositeswith
application-dictated stiffness and hardness.Our study combining theoretical and experimentalmethods exemplifies the strength of
this approach for the case of the elastic properties of AlN/CrNsuperlattices that were deposited by reactive radio-frequencymagnetron sputtering with a bilayer period of 4 nm. Importantly,
CrN stabilizes AlNin ametastable B1 (rock salt) cubic phase only in the formof a layer that is very thin, up to a fewnanometers.Due to the fact thatB1-AlNcrystals do not exist as
bulkmaterials, experimental data
for this phase are not available. Therefore, quantum-mechanical calculations have been applied to simulate an AlN/CrNsuperlatticewith a similar bilayer period.The ab initio predicted
Youngʼs modulus (428GPa) along the [001] direction is in excellent agreement with measured nano-indentation values (408±32 GPa).Aiming at a future rapid high-throughputmaterials design of
superlattices,we have also tested predictions obtained within linear-elasticity continuum modeling using elastic properties of B1-CrNand B1-AlN phases as input.Using single-crystal elastic
constants fromab initio calculations for both phases, we demonstrate the reliability of this approach to design nano-patterned
coherent superlatticeswith unprecedented and potentially superior properties.
How to do Combinatorial Alloy Design by Laser Additive Manufacturing ?
In some projects we have used laser additive manufacturing as a combinatorial method for synthesizing microstructurally and compositionally piecewise graded bulk alloys. For the case of ultrahard tool steels we fabricated blocks consisting of a sequence of ~500 µm thick tool steel layers, each with different chemical composition, by laser metal deposition where alloy powders were deposited layer-wise on a substrate. The reference materials are a Cr-Mo-V hot working tool steel and a Ni-based maraging steel. The layers between them consist of corresponding blends of the two materials with varying composition from layer to layer (alloy volume fractions 80:20, 60:40, 40:60, 20:80). The bulk alloy was hot rolled and heat treated. Subsequently each layer was characterized for microstructure, chemical composition and mechanical properties using electron back scatter diffraction, tensile testing and indentation. The approach is an efficient high-throughput method enabling rapid probing of novel compositional alloy blends. It can be applied for finding new alloys both, by laser additive manufacturing and for laser additive manufacturing. For the tool steel blends synthesized here we observe that the Cr-Mo-V tool steel, when mixed with the Ni-base maraging steel, can be continuously tuned for a strength-ductility profile in the range of 800 – 1650 MPa strength and 15-25% tensile elongation.
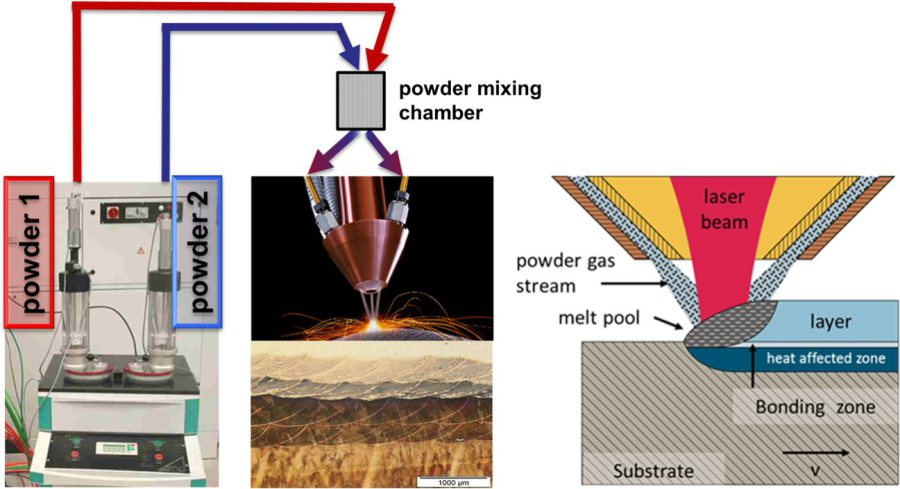 Combinatorial Alloy Design by Laser Additive Manufacturing .
Combinatorial Alloy Design by Laser Additive Manufacturing .
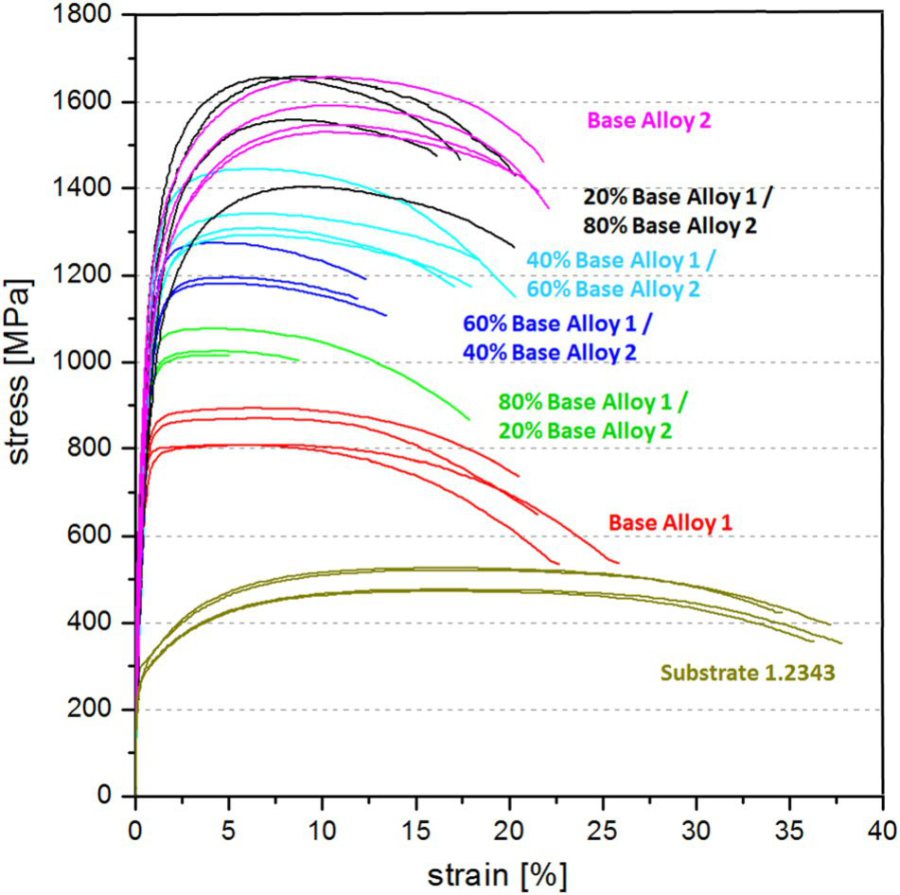 Combinatorial Alloy Design by Laser Additive Manufacturing. Tensile test results conducted on the deformed and heat treated samples.
Combinatorial Alloy Design by Laser Additive Manufacturing. Tensile test results conducted on the deformed and heat treated samples.
This work presents a new approach to rapid bulk alloy screening combining two metallurgical research fields, namely, Laser Additive Manufacturing (LAM) and combinatorial bulk alloy design. As an example system we investigate different alloy blends of two types of tool steels, a Cr–Mo–V hot working tool steel and a Ni-based maraging steel. Several earlier papers have also explored different kinds of pathways for synthesizing gradient alloy combinations by using LAM methods. Although some of these earlier methods did not impose additional thermal mechanical processing after the deposition of alloys with compositional gradients, these studies revealed the capability of using additive manufacturing to explore combinatorial bulk samples.
2016-steel research laser additive manuf[...]
PDF-Dokument [3.6 MB]
Acta Materialia 60 (2012) 4950-4959
Acta Materialia 60 (2012) 4950 combinato[...]
PDF-Dokument [1.3 MB]
We introduce a new experimental approach to the compositional and thermo-mechanical design and rapid maturation of bulk structural materials. This method, termed rapid alloy prototyping
(RAP), is based on semi-continuous high throughput bulk casting, rolling, heat treatment and sample preparation techniques. 45 Material conditions, i.e. 5 alloys with systematically varied
compositions, each modified by 9 different ageing treatments, were produced and investigated within 35 h. This accelerated screening of the tensile, hardness
and microstructural properties as a function of chemical and thermo-mechanical parameters allows the highly efficient and knowledge-based design of bulk structural alloys. The efficiency of the
approach was demonstrated on a group of Fe–30Mn–1.2C–xAl steels which exhibit a wide spectrum of structural and mechanical characteristics, depending on the respective Al concentration. High
amounts of Al addition (>8 wt.%) resulted in pronounced strengthening, while low concentrations (<2 wt.%) led to embrittlement of the material during ageing.
Acta Materialia 70 (2014) 92
S. Sandlöbes, Z. Pei, M. Friak, L.-F. Zhu, F. Wang, S. Zaefferer, D. Raabe, J. Neugebauer
Acta Materialia 70 (2014) 92 solid solut[...]
PDF-Dokument [891.1 KB]
The I1 intrinsic stacking fault energy (I1 SFE) serves as an alloy design parameter for ductilizing Mg alloys. In view of this effect we have conducted quantum–mechanical calculations for Mg15X solid-solution crystals (X = Dy, Er, Gd, Ho, Lu, Sc, Tb, Tm, Nd, Pr, Be, Ti, Zr, Zn, Tc, Re, Co, Ru, Os, Tl). We find that Y, Sc and all studied lanthanides reduce the I1 SFE and render hexagonal closed-packed (hcp) and double hcp phases thermodynamically, structurally and elastically similar. Synthesis, experimental testing and characterization of some of the predicted key alloys (Mg–3Ho, Mg–3Er, Mg–3Tb, Mg–3Dy) indeed confirm reduced I1 SFEs and significantly improved room-temperature ductility by up to 4–5 times relative to pure Mg, a finding that is attributed to the higher activity of non-basal dislocation slip.
Acta Materialia 57 (2009) 69
W.A. Counts, M. Friak, D. Raabe, J. Neugebauer
Using ab initio calculations in designing bcc Mg–Li alloys for ultra-lightweight applications
Acta Materialia 57 (2009) 69-76 ab initi[...]
PDF-Dokument [499.7 KB]
Ab initio calculations are becoming increasingly useful to engineers interested in designing new alloys, because these calculations are able to accurately predict basic material properties
only knowing the atomic composition of the material. In this paper, single crystal elastic constants of 11 bcc Mg–Li alloys are calculated using density functional theory (DFT) and compared with
available experimental
data. Based on DFT determined properties, engineering parameters such as the ratio of bulk modulus over shear modulus (B/G) and the ratio of Young’s modulus over mass density (Y/q) are
calculated. Analysis of B/G and Y/q shows that bcc Mg–Li alloys with 30–50 at.% Li offer the most potential as lightweight structural material. Compared with fcc Al–Li alloys, bcc Mg–Li alloys
have a lower B/G ratio, but a comparable Y/q ratio. An Ashby map containing Y/q vs B/G shows that it is not possible to increase both Y/q and B/G by changing
only the composition of a binary alloy.
Acta Materialia 60 (2012) 6025-6032
Acta Materialia 60 (2012) 6025–6032 Comb[...]
PDF-Dokument [887.1 KB]
We have studied experimentally and theoretically the influence of C and Mn content on the Young’s modulus of Fe–Mn–C alloys. Combinatorial thin film and bulk samples were characterized regarding their structure, texture and Young’s modulus. The following chemical composition range was investigated: 1.5–3.0 at.% C, 28.0–37.5 at.% Mn and 60.6–69.8 at.% Fe. The experimental lattice parameters change marginally within 3.597–3.614A ˚ with the addition of C and are consistent with ab initio calculations. The Young’s modulus data are in the range of 185 ± 12–251 ± 59 GPa for the bulk samples and the thin film, respectively. C has no significant effect on the Young’s modulus of these alloys within the composition range studied here. The ab initio calculations are 15–22% larger than the average Young’s modulus values of the as-deposited and polished thin film at 3 at.% C. The comparison of thin film and bulk samples results reveals similar elastic properties for equivalent compositions, indicating that the applied research strategy consisting of the combinatorial thin film approach in conjunction with ab initio calculations is useful to study the composition dependence of the structure and elastic properties of Fe–Mn–C alloys. The very good agreement between the presented calculations and the experimentally determined lattice parameters and Young’s modulus values implies that the here-adopted simulation strategy yields a reliable description of carbon in Fe– Mn alloys, important for future alloy design.
Acta Materialia 85 (2015) 53
Computationally efficient and quantitatively accurate multiscale simulation of solid-solution strengthening by ab initio calculation
Duancheng Ma,Martin Friak, Johann von Pezold, Dierk Raabea and Jörg Neugebauer
Acta Mater 85 (2015) 53 Ma et al - solid[...]
PDF-Dokument [2.3 MB]
We propose an approach for the computationally efficient and quantitatively accurate prediction of solid-solution strengthening. It combines the 2-D Peierls–Nabarro model and a recently developed solid-solution strengthening model. Solid-solution strengthening is examined with Al–Mg and Al–Li as representative alloy systems, demonstrating a good agreement between theory and experiments within the temperature range in which the dislocation motion is overdamped. Through a parametric study, two guideline maps of the misfit parameters against (i) the critical resolved shear stress, s0, at 0 K and (ii) the energy barrier, DEb, against dislocation motion in a solid solution with randomly distributed solute atoms are created. With these two guideline maps, s0 at finite temperatures is predicted for other Al binary systems, and compared with available experiments, achieving good agreement.
Acta Materialia 98 (2015) 367-376
Ab initio study of compositional trends in solid solution strengthening in metals with low Peierls stresses
Duancheng Ma, Martin Friák, Johann von Pezold, Jörg Neugebauer, Dierk Raabe
Acta Materialia 2015 ab initio solution [...]
PDF-Dokument [298.4 KB]
We identify and analyze general trends governing solid solution strengthening in binary alloys containing solutes across the Periodic table using quantum-mechanical calculations. Here we
present calculations for the model system of Al binary solid solutions. The identified trends originate from an approximately
parabolic dependence of two strengthening parameters to quantitatively predict the solid solution strengthening effect, i.e. the volume and slip misfit parameters. The volume misfit parameter
shows a minimum (concave-up behavior) as a function of the solute element group number in the periodic table, whereas the slip misfit parameter shows a maximum (concave-down behavior). By
analyzing reported data, a similar trend is also found in Ni and Mg (basal slip) binary systems. Hence, these two strengthening parameters are strongly anti-correlated, which can be understood
in terms of the Fermi level shift in
the framework of free electron model. The chemical trends identified in this study enable a rapid and efficient identification of the solutes that provide optimum solid–solution strengthening.
The approach described here may thus serve as basis for ab initio guided metallurgical materials design.